Ren Ze, Niu Decao, Ma Panpan, Wang Ying, Wang Zhaomin, Fu Hua, Elser James J
State Key Laboratory of Grassland Agro-ecosystems, Key Laboratory of Grassland Livestock Industry Innovation, Ministry of Agriculture and Rural Affairs, Engineering Research Center of Grassland Industry, Ministry of Education, College of Pastoral Agriculture Science and Technology, Lanzhou University, Lanzhou, China.
Flathead Lake Biological Station, University of Montana, Polson, MT, United States.
Front Microbiol. 2020 Jun 5;11:1021. doi: 10.3389/fmicb.2020.01021. eCollection 2020.
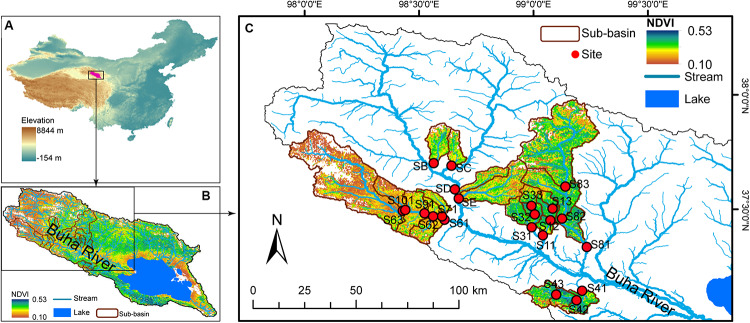
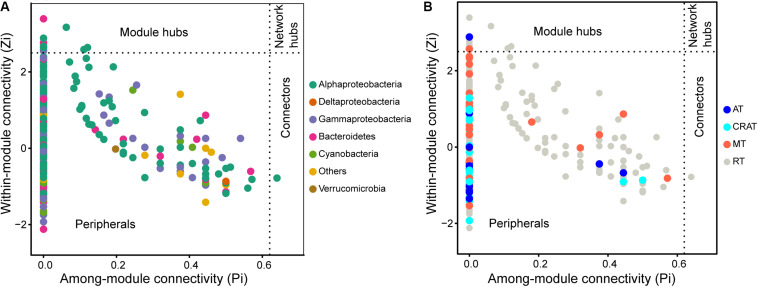
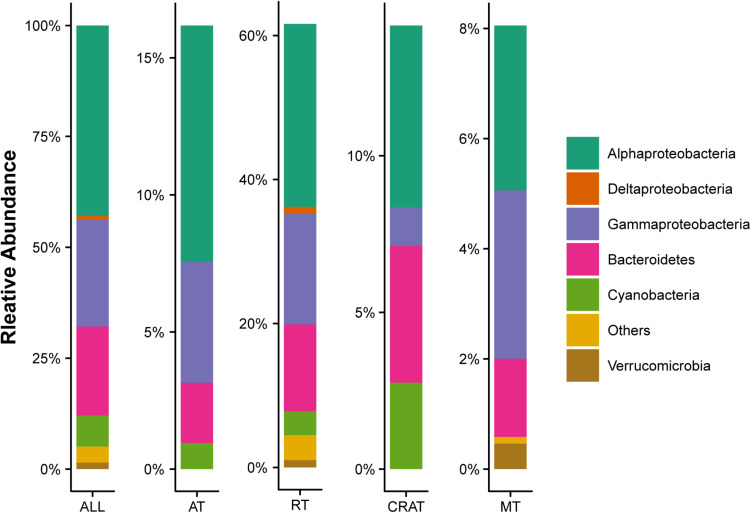
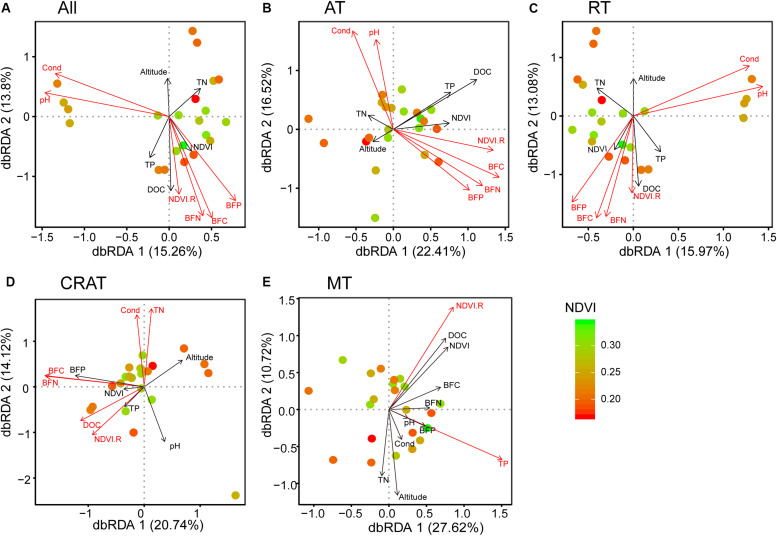
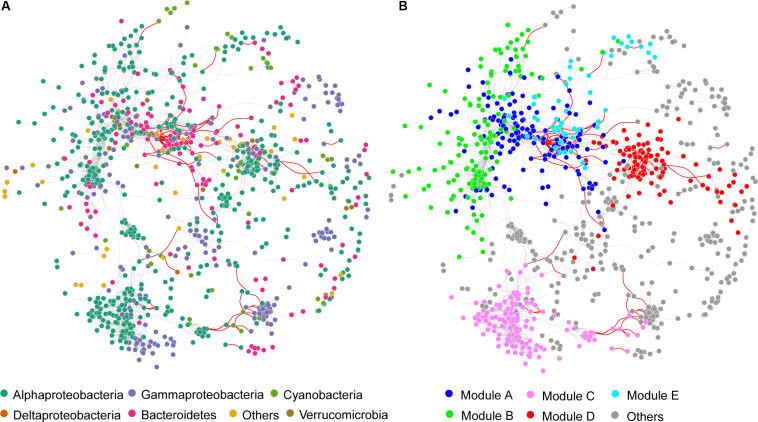
Grassland is among the largest terrestrial biomes and is experiencing serious degradation, especially on the Qinghai-Tibet Plateau (QTP). However, the influences of grassland degradation on microbial communities in stream biofilms are largely unknown. Using 16S rRNA gene sequencing, we investigated the bacterial communities in stream biofilms in sub-basins with different grassland status in the Qinghai Lake watershed. Grassland status in the sub-basins was quantified using the normalized difference vegetation index (NDVI). Proteobacteria, Bacteroidetes, Cyanobacteria, and Verrucomicrobia were the dominant bacterial phyla. OTUs, 7,050, were detected in total, within which 19 were abundant taxa, and 6,922 were rare taxa. Chao 1, the number of observed OTUs, and phylogenetic diversity had positive correlations with carbon (C), nitrogen (N), and/or phosphorus (P) in biofilms . The variation of bacterial communities in stream biofilms was closely associated with the rate of change in NDVI, pH, conductivity, as well as C, N, P, contents and C:N ratio of the biofilms. Abundant subcommunities were more influenced by environmental variables relative to the whole community and to rare subcommunities. These results suggest that the history of grassland degradation (indicated as the rate of change in NDVI) influences bacterial communities in stream biofilms. Moreover, the bacterial community network showed high modularity with five major modules (>50 nodes) that responded differently to environmental variables. According to the module structure, only one module connector and 12 module hubs were identified, suggesting high fragmentation of the network and considerable independence of the modules. Most of the keystone taxa were rare taxa, consistent with fragmentation of the network and with adverse consequences for bacterial community integrity and function in the biofilms. By documenting the properties of bacterial communities in stream biofilms in a degrading grassland watershed, our study adds to our knowledge of the potential influences of grassland degradation on aquatic ecosystems.
草原是最大的陆地生物群落之一,并且正经历着严重的退化,尤其是在青藏高原(QTP)。然而,草原退化对溪流生物膜中微生物群落的影响在很大程度上尚不清楚。我们利用16S rRNA基因测序技术,研究了青海湖流域不同草原状况子流域溪流生物膜中的细菌群落。利用归一化植被指数(NDVI)对各子流域的草原状况进行了量化。变形菌门、拟杆菌门、蓝细菌门和疣微菌门是主要的细菌门类。总共检测到7050个操作分类单元(OTU),其中19个是丰富分类群,6922个是稀有分类群。Chao 1、观察到的OTU数量和系统发育多样性与生物膜中的碳(C)、氮(N)和/或磷(P)呈正相关。溪流生物膜中细菌群落的变化与NDVI、pH值、电导率的变化率以及生物膜中的C、N、P含量和C:N比密切相关。相对于整个群落和稀有亚群落,丰富亚群落受环境变量的影响更大。这些结果表明,草原退化历史(以NDVI变化率表示)影响溪流生物膜中的细菌群落。此外,细菌群落网络显示出高度的模块化,有五个主要模块(>50个节点)对环境变量有不同的响应。根据模块结构,仅识别出一个模块连接点和12个模块枢纽,表明网络高度碎片化且模块具有相当大的独立性。大多数关键分类群是稀有分类群,这与网络的碎片化以及对生物膜中细菌群落完整性和功能的不利影响相一致。通过记录退化草原流域溪流生物膜中细菌群落的特性,我们的研究增加了我们对草原退化对水生生态系统潜在影响的认识。