Tao Wen-Yuan, Yu Lin-Jie, Jiang Su, Cao Xiang, Chen Jian, Bao Xin-Yu, Li Fei, Xu Yun, Zhu Xiao-Lei
Department of Neurology, Drum Tower Hospital, Medical School of Nanjing University; The State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University; Jiangsu Key Laboratory for Molecular Medicine, Nanjing, Jiangsu Province, China.
Taizhou People's Hospital, Taizhou, Jiangsu Province, China.
Neural Regen Res. 2020 Dec;15(12):2296-2305. doi: 10.4103/1673-5374.285006.
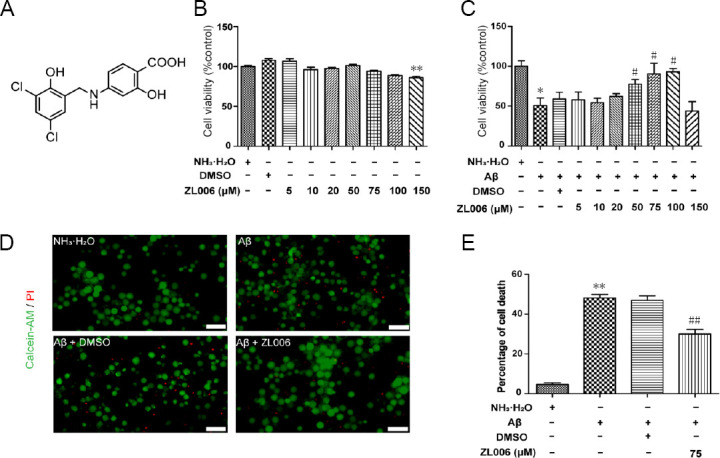
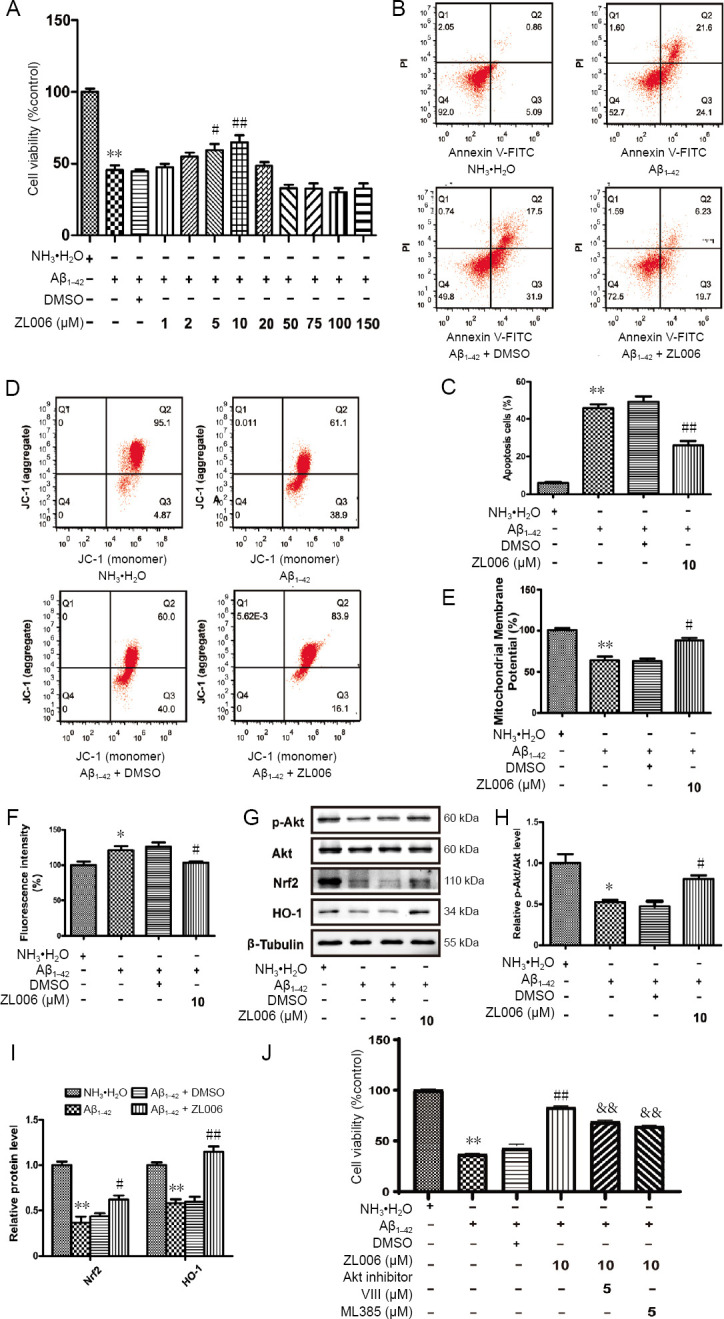
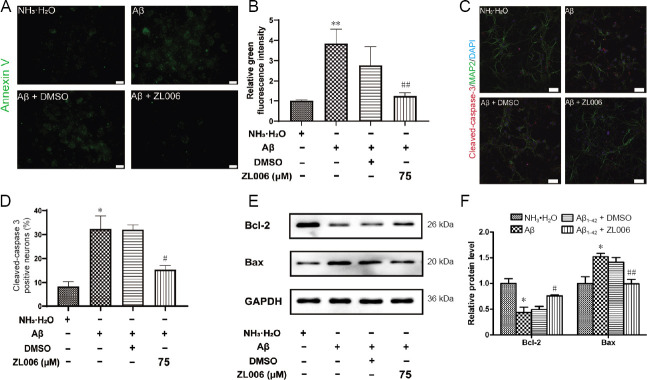
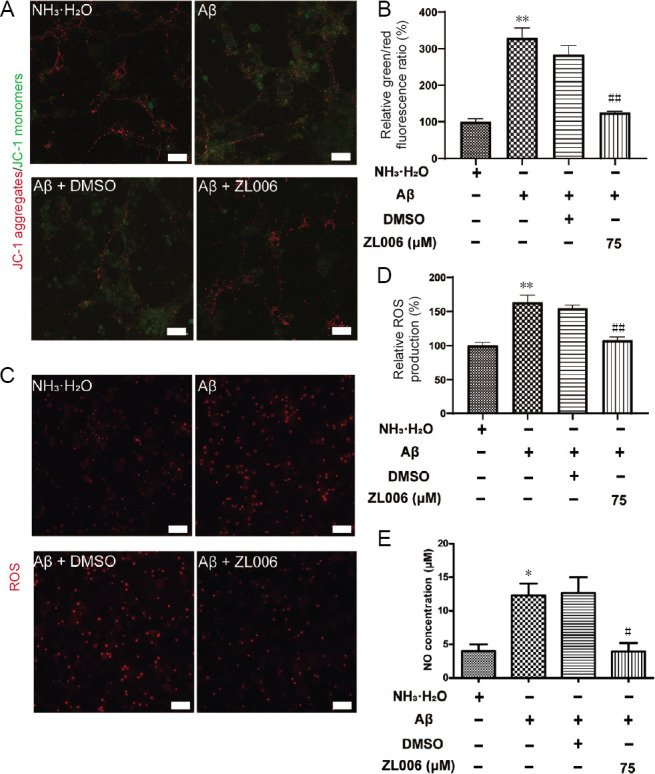
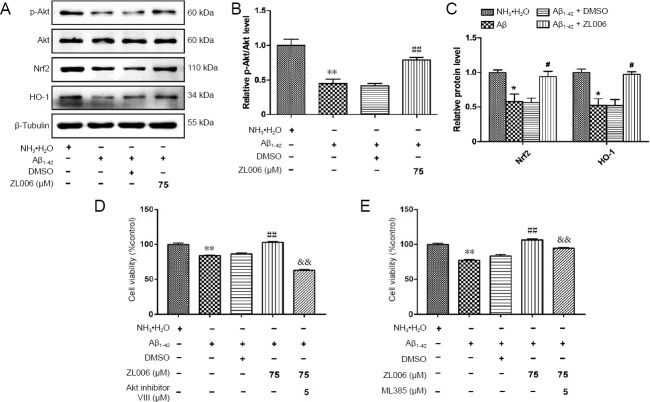
Amyloid beta (Aβ)-induced neurotoxicity and oxidative stress plays an important role in the pathogenesis of Alzheimer's disease (AD). ZL006 is shown to reduce over-produced nitric oxide and oxidative stress in ischemic stroke by interrupting the interaction of neuronal nitric oxide synthase and postsynaptic density protein 95. However, few studies are reported on the role of ZL006 in AD. To investigate whether ZL006 exerted neuroprotective effects in AD, we used Aβ to treat primary cortical neurons and N2a neuroblastoma cells as an in vitro model of AD. Cortical neurons were incubated with ZL006 or dimethyl sulfoxide for 2 hours and treated with Aβ or NH•HO for another 24 hours. The results of cell counting Kit-8 (CCK-8) assay and calcein-acetoxymethylester/propidium iodide staining showed that ZL006 pretreatment rescued the neuronal death induced by Aβ. Fluorescence and western blot assay were used to detect oxidative stress and apoptosis-related proteins in each group of cells. Results showed that ZL006 pretreatment decreased neuronal apoptosis and oxidative stress induced by Aβ. The results of CCK8 assay showed that inhibition of Akt or NF-E2-related factor 2 (Nrf2) in cortical neurons abolished the protective effects of ZL006. Moreover, similar results were also observed in N2a neuroblastoma cells. ZL006 inhibited N2a cell death and oxidative stress induced by Aβ, while inhibition of Akt or Nrf2 abolished the protective effect of ZL006. These results demonstrated that ZL006 reduced Aβ-induced neuronal damage and oxidative stress, and the mechanisms might be associated with the activation of Akt/Nrf2/heme oxygenase-1 signaling pathways.
β-淀粉样蛋白(Aβ)诱导的神经毒性和氧化应激在阿尔茨海默病(AD)的发病机制中起重要作用。已表明ZL006通过中断神经元型一氧化氮合酶与突触后致密蛋白95的相互作用来减少缺血性卒中中过量产生的一氧化氮和氧化应激。然而,关于ZL006在AD中的作用的报道很少。为了研究ZL006是否在AD中发挥神经保护作用,我们使用Aβ处理原代皮质神经元和N2a神经母细胞瘤细胞作为AD的体外模型。将皮质神经元与ZL006或二甲基亚砜孵育2小时,然后再用Aβ或NH•HO处理24小时。细胞计数试剂盒-8(CCK-8)检测和钙黄绿素-乙酰氧基甲酯/碘化丙啶染色结果表明,ZL006预处理可挽救Aβ诱导的神经元死亡。采用荧光和蛋白质印迹法检测每组细胞中的氧化应激和凋亡相关蛋白。结果表明,ZL006预处理可降低Aβ诱导的神经元凋亡和氧化应激。CCK8检测结果表明,抑制皮质神经元中的Akt或核因子E2相关因子2(Nrf2)可消除ZL006的保护作用。此外,在N2a神经母细胞瘤细胞中也观察到了类似的结果。ZL006抑制Aβ诱导的N2a细胞死亡和氧化应激,而抑制Akt或Nrf2则消除了ZL006的保护作用。这些结果表明,ZL006可减轻Aβ诱导的神经元损伤和氧化应激,其机制可能与Akt/Nrf2/血红素加氧酶-1信号通路的激活有关。