Gillings School of Global Public Health (A.R.B., H.M.H., R.G., M.G., C.J.H., A.A.S., E.A.W., K.E.N., C.L.A.), University of North Carolina at Chapel Hill.
Cardiovascular Health Research Unit, Department of Medicine (C.M.S.), University of Washington, Seattle.xs.
Circ Genom Precis Med. 2020 Aug;13(4):e002680. doi: 10.1161/CIRCGEN.119.002680. Epub 2020 Jun 30.
We examined how expanding electrocardiographic trait genome-wide association studies to include ancestrally diverse populations, prioritize more precise phenotypic measures, and evaluate evidence for shared genetic effects enabled the detection and characterization of loci.
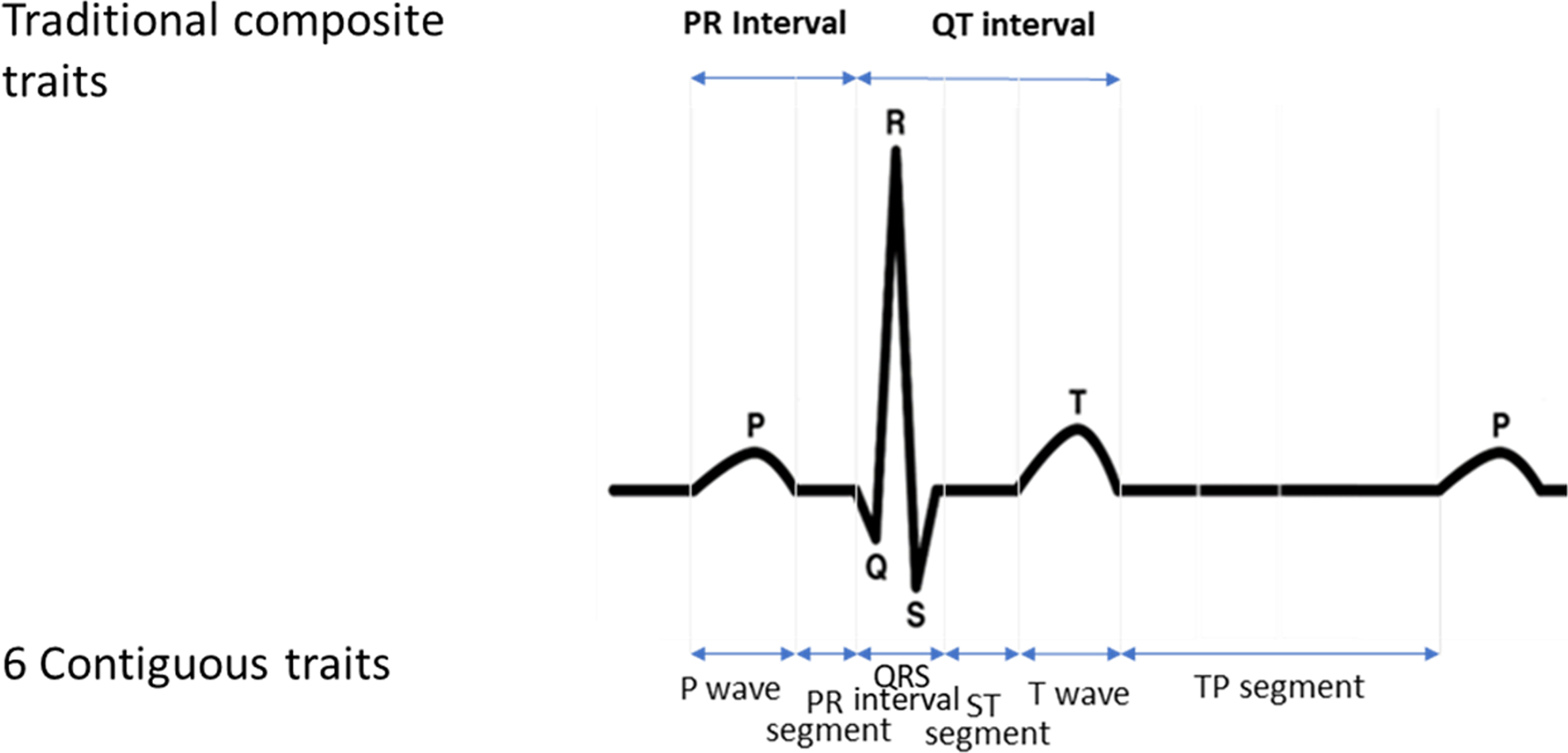
We decomposed 10 seconds, 12-lead electrocardiograms from 34 668 multi-ethnic participants (15% Black; 30% Hispanic/Latino) into 6 contiguous, physiologically distinct (P wave, PR segment, QRS interval, ST segment, T wave, and TP segment) and 2 composite, conventional (PR interval and QT interval) interval scale traits and conducted multivariable-adjusted, trait-specific univariate genome-wide association studies using 1000-G imputed single-nucleotide polymorphisms. Evidence of shared genetic effects was evaluated by aggregating meta-analyzed univariate results across the 6 continuous electrocardiographic traits using the combined phenotype adaptive sum of powered scores test.
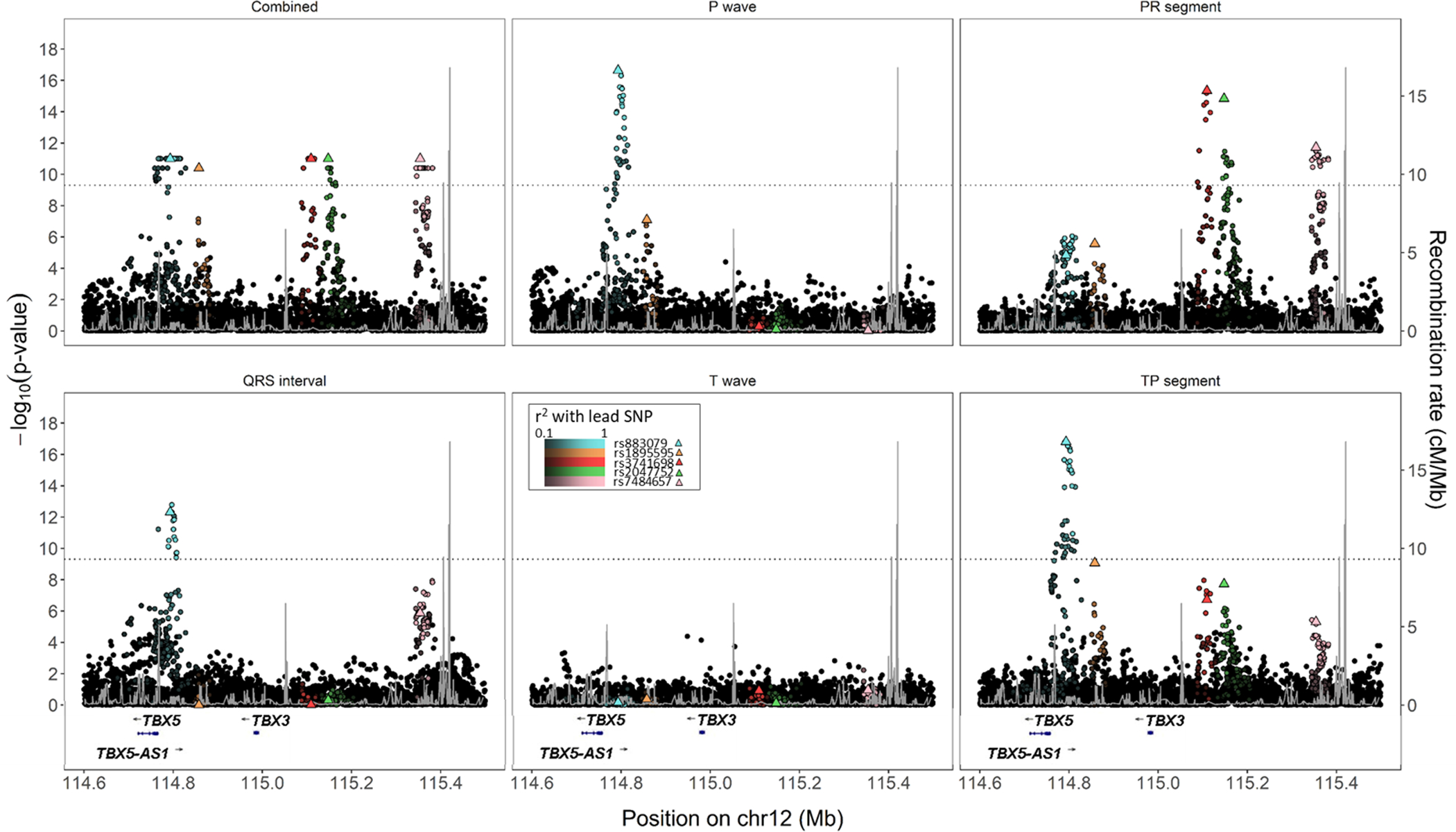
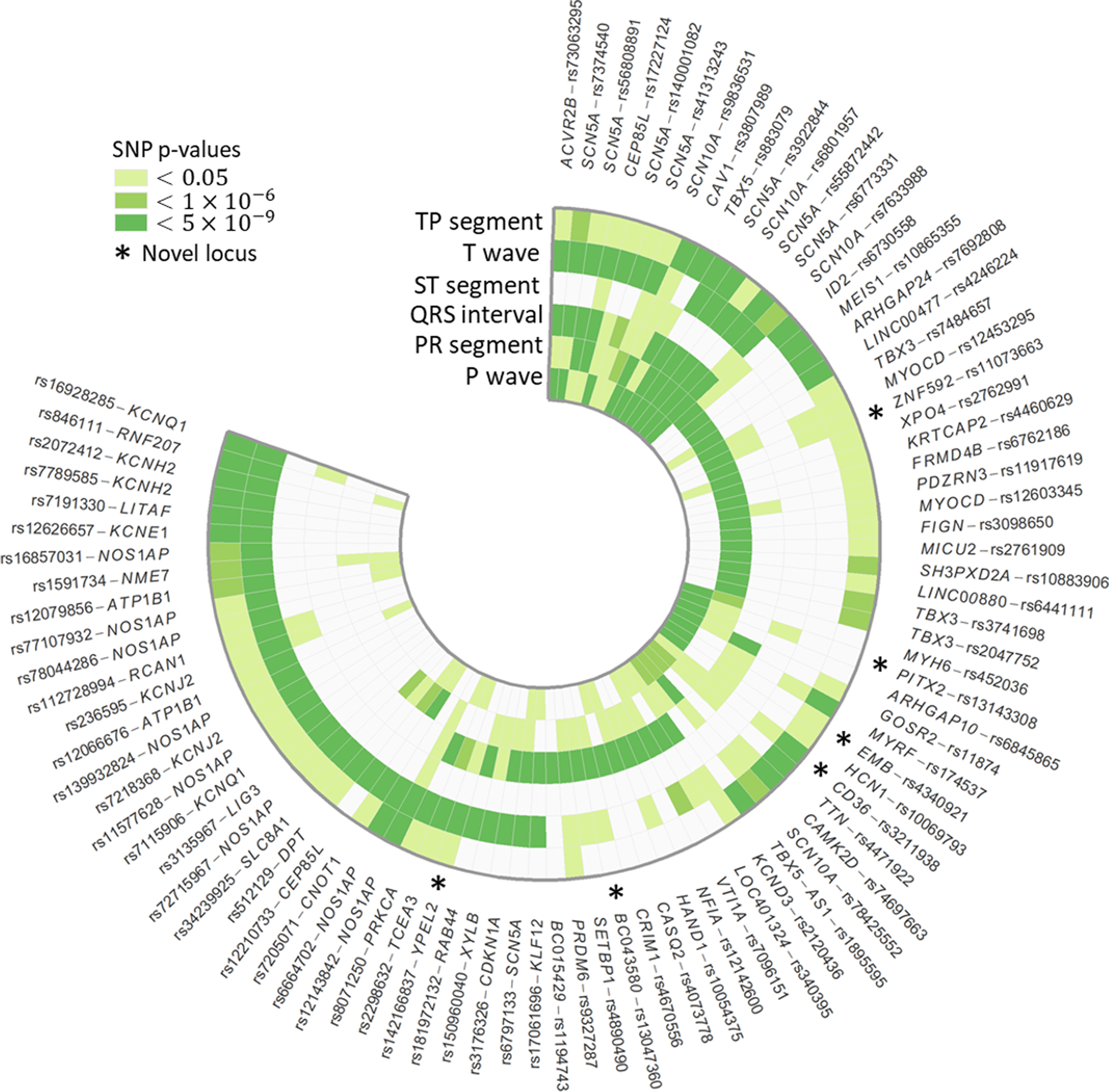
We identified 6 novels (, and ) and 87 known loci (adaptive sum of powered score test <5×10). Lead single-nucleotide polymorphism rs3211938 at was common in Blacks (minor allele frequency=10%), near monomorphic in European Americans, and had effects on the QT interval and TP segment that ranked among the largest reported to date for common variants. The other 5 novel loci were observed when evaluating the contiguous but not the composite electrocardiographic traits. Combined phenotype testing did not identify novel electrocardiographic loci unapparent using traditional univariate approaches, although this approach did assist with the characterization of known loci.
Despite including one-third as many participants as published electrocardiographic trait genome-wide association studies, our study identified 6 novel loci, emphasizing the importance of ancestral diversity and phenotype resolution in this era of ever-growing genome-wide association studies.
我们研究了如何通过扩大心电图特征的全基因组关联研究范围,纳入更多具有不同祖先的人群,优先考虑更精确的表型测量,并评估遗传效应的共享证据,来检测和描述新的遗传位点。
我们将来自 34668 名多民族参与者(15%为黑人,30%为西班牙裔/拉丁裔)的 10 秒 12 导联心电图分解为 6 个连续的、具有生理差异的(P 波、PR 段、QRS 间隔、ST 段、T 波和 TP 段)和 2 个复合的、常规的(PR 间隔和 QT 间隔)间隔尺度特征,并使用 1000G imputed 单核苷酸多态性进行多变量调整后的特征特异性单变量全基因组关联研究。通过对 6 个连续心电图特征的元分析单变量结果进行汇总,使用合并表型自适应加权得分检验来评估遗传效应的共享证据。
我们鉴定出 6 个新的(、和)和 87 个已知的(自适应加权得分检验<5×10)位点。位于的 lead 单核苷酸多态性 rs3211938 在黑人中较为常见(次要等位基因频率=10%),在欧洲裔美国人中几乎是单态的,对 QT 间隔和 TP 段的影响是迄今为止报道的常见变异中最大的之一。当评估连续的而不是复合的心电图特征时,观察到其他 5 个新的遗传位点。尽管这种方法有助于描述已知的遗传位点,但综合表型检验并未发现传统单变量方法中未发现的新的心电图遗传位点。
尽管纳入的参与者人数仅为已发表的心电图特征全基因组关联研究的三分之一,但我们的研究鉴定出了 6 个新的遗传位点,这强调了在全基因组关联研究不断增长的时代,祖先多样性和表型分辨率的重要性。