Institute for Memory Impairments and Neurological Disorders, University of California, Irvine, Irvine, CA, 92697, USA.
Department of Neurobiology and Behavior, University of California, Irvine, Irvine, CA, 92697, USA.
Alzheimers Res Ther. 2020 Jul 6;12(1):80. doi: 10.1186/s13195-020-00649-8.
Cathepsin D (CatD) is a lysosomal protease that degrades both the amyloid β-protein (Aβ) and the microtubule-associated protein, tau, and has been genetically linked to late-onset Alzheimer disease (AD). Here, we sought to examine the consequences of genetic deletion of CatD on Aβ proteostasis in vivo and to more completely characterize the degradation of Aβ42 and Aβ40 by CatD.
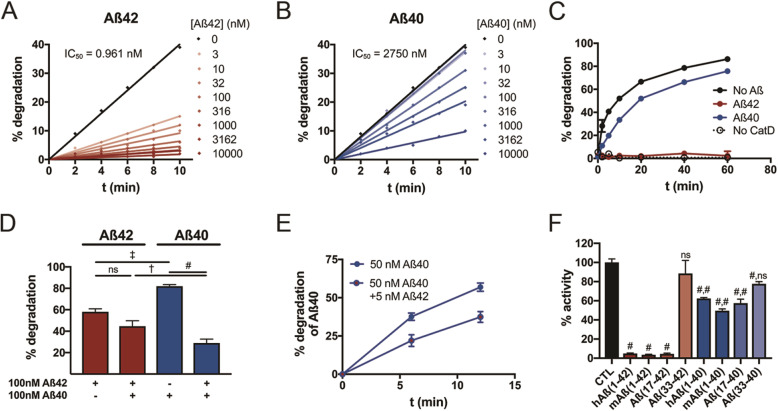
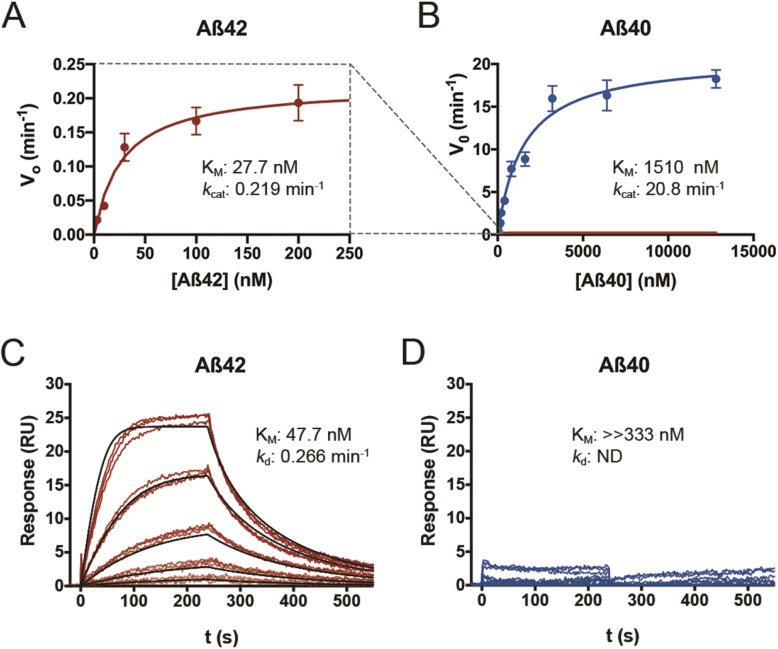
We quantified Aβ degradation rates and levels of endogenous Aβ42 and Aβ40 in the brains of CatD-null (CatD-KO), heterozygous null (CatD-HET), and wild-type (WT) control mice. CatD-KO mice die by ~ 4 weeks of age, so tissues from younger mice, as well as embryonic neuronal cultures, were investigated. Enzymological assays and surface plasmon resonance were employed to quantify the kinetic parameters (K, k) of CatD-mediated degradation of monomeric human Aβ42 vs. Aβ40, and the degradation of aggregated Aβ42 species was also characterized. Competitive inhibition assays were used to interrogate the relative inhibition of full-length human and mouse Aβ42 and Aβ40, as well as corresponding p3 fragments.
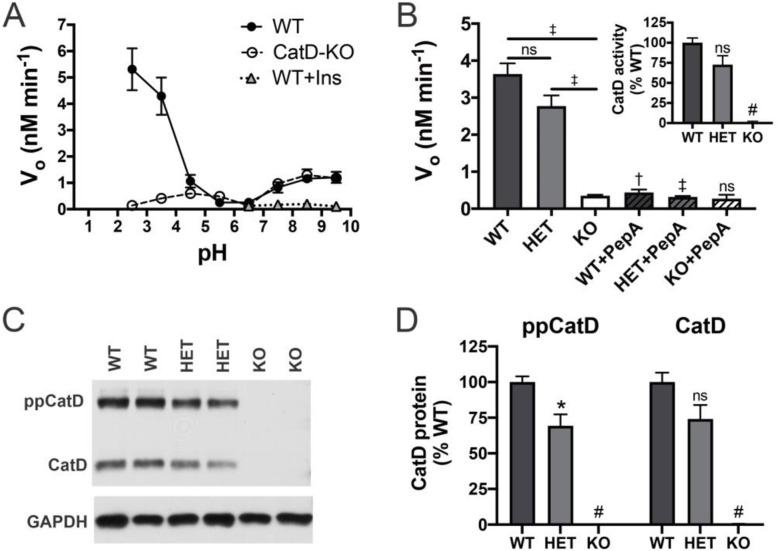
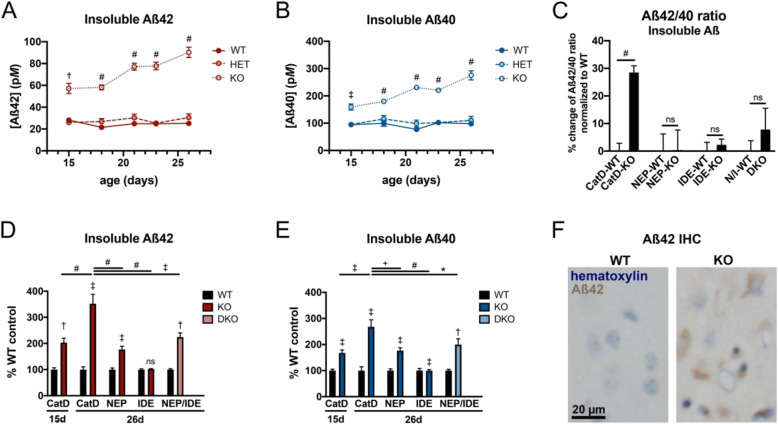
Genetic deletion of CatD resulted in 3- to 4-fold increases in insoluble, endogenous cerebral Aβ42 and Aβ40, exceeding the increases produced by deletion of an insulin-degrading enzyme, neprilysin or both, together with readily detectable intralysosomal deposits of endogenous Aβ42-all by 3 weeks of age. Quite significantly, CatD-KO mice exhibited ~ 30% increases in Aβ42/40 ratios, comparable to those induced by presenilin mutations. Mechanistically, the perturbed Aβ42/40 ratios were attributable to pronounced differences in the kinetics of degradation of Aβ42 vis-à-vis Aβ40. Specifically, Aβ42 shows a low-nanomolar affinity for CatD, along with an exceptionally slow turnover rate that, together, renders Aβ42 a highly potent competitive inhibitor of CatD. Notably, the marked differences in the processing of Aβ42 vs. Aβ40 also extend to p3 fragments ending at positions 42 vs. 40.
Our findings identify CatD as the principal intracellular Aβ-degrading protease identified to date, one that regulates Aβ42/40 ratios via differential degradation of Aβ42 vs. Aβ40. The finding that Aβ42 is a potent competitive inhibitor of CatD suggests a possible mechanistic link between elevations in Aβ42 and downstream pathological sequelae in AD.
组织蛋白酶 D(CatD)是一种溶酶体蛋白酶,可降解淀粉样β-蛋白(Aβ)和微管相关蛋白,并且已在遗传上与晚发性阿尔茨海默病(AD)相关。在这里,我们试图研究 CatD 基因缺失对体内 Aβ 稳定性的影响,并更全面地描述 CatD 对 Aβ42 和 Aβ40 的降解。
我们定量了 CatD 缺失(CatD-KO)、杂合缺失(CatD-HET)和野生型(WT)对照小鼠脑中 Aβ 降解率以及内源性 Aβ42 和 Aβ40 的水平。CatD-KO 小鼠在大约 4 周龄时死亡,因此研究了年龄较小的小鼠以及胚胎神经元培养物中的组织。酶学测定和表面等离子体共振用于量化单体人 Aβ42 与 Aβ40 之间 CatD 介导的降解的动力学参数(K,k),并且还表征了聚集的 Aβ42 物种的降解。竞争性抑制测定用于研究全长人 Aβ42 和 Aβ40 以及相应的 p3 片段的相对抑制作用。
CatD 的基因缺失导致可溶性、内源性大脑 Aβ42 和 Aβ40 增加了 3-4 倍,超过了胰岛素降解酶、neprilysin 或两者共同缺失以及 3 周龄时可检测到的内溶酶体 Aβ42 沉积所产生的增加。非常显著的是,CatD-KO 小鼠的 Aβ42/40 比值增加了约 30%,与早老素突变诱导的比值相当。从机制上讲,Aβ42/40 比值的紊乱归因于 Aβ42 与 Aβ40 降解动力学的明显差异。具体而言,Aβ42 对 CatD 具有低纳摩尔亲和力,并且周转率非常缓慢,这使得 Aβ42 成为 CatD 的高度有效的竞争性抑制剂。值得注意的是,Aβ42 与 Aβ40 相比的处理差异也扩展到了位置 42 与 40 处的 p3 片段。
我们的研究结果确定 CatD 是迄今为止鉴定的主要细胞内 Aβ 降解蛋白酶,它通过 Aβ42 与 Aβ40 的差异降解来调节 Aβ42/40 比值。Aβ42 是 CatD 的有效竞争性抑制剂的发现表明,AD 中 Aβ42 升高与下游病理后果之间可能存在机制联系。