Soybean Research Institute, Nanjing Agricultural University, Nanjing 210095, China.
MOA National Center for Soybean Improvement, Nanjing Agricultural University, Nanjing 210095, China.
Int J Mol Sci. 2020 Jul 8;21(14):4830. doi: 10.3390/ijms21144830.
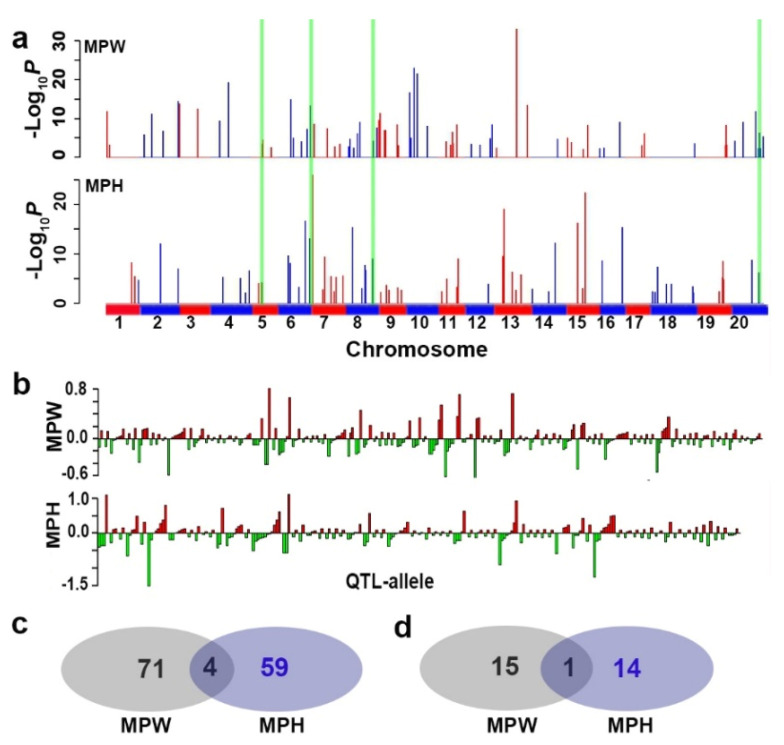
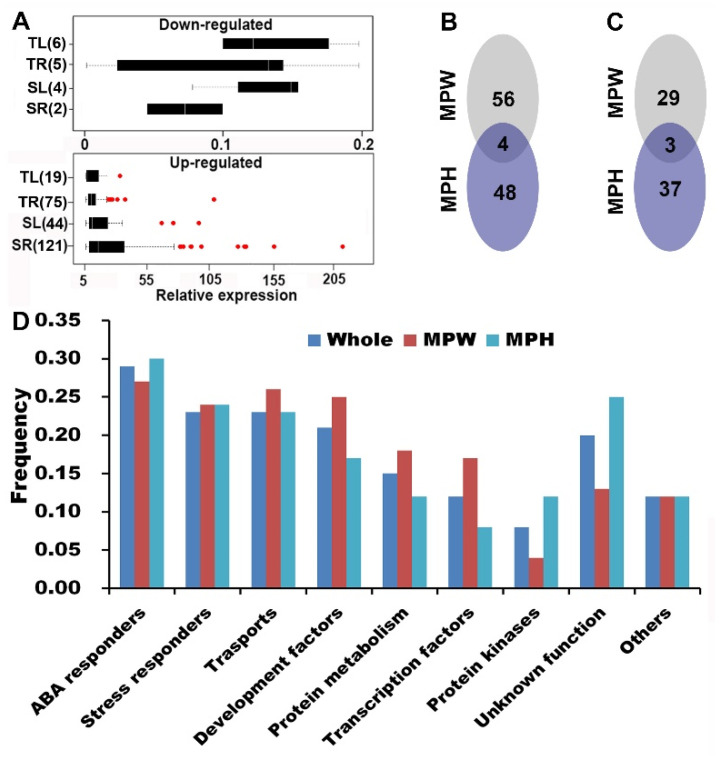
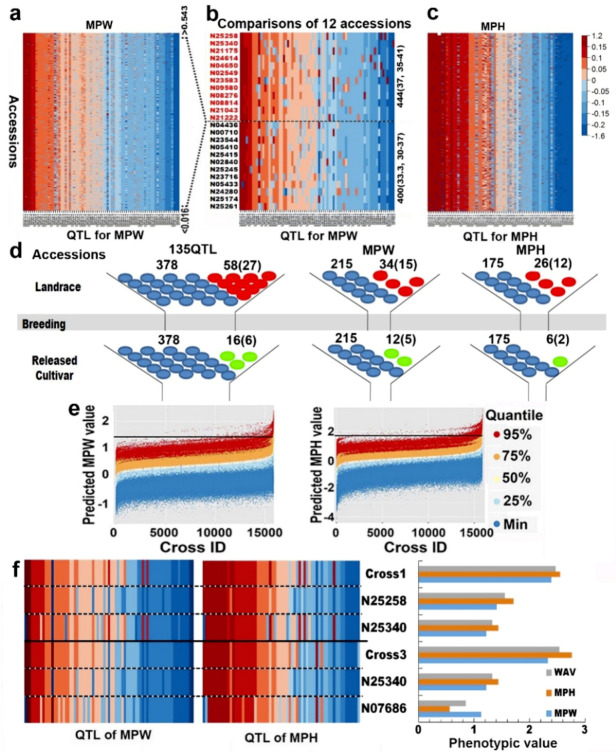
Drought is one of the most important factors affecting plant growth and productivity. The previous results on drought tolerance (DT) genetic system in soybean indicated a complex of genes not only few ones were involved in the trait. This study is featured with a relatively thorough identification of QTL-allele/candidate-gene system using an efficient restricted two-stage multi-locus multi-allele genome-wide association study, on two comprehensive DT indicators, membership index values of relative plant weight (MPW) and height (MPH), instead of a single biological characteristic, in a large sample (564 accessions) of the Chinese cultivated soybean population (CCSP). Based on 24,694 multi-allele markers, 75 and 64 QTL with 261 and 207 alleles (2-12/locus) were detected for MPW and MPH, explaining 54.7% and 47.1% of phenotypic variance, respectively. The detected QTL-alleles were organized into a QTL-allele matrix for each indicator, indicating DT is a super-trait conferred by two (even more) QTL-allele systems of sub-traits. Each CCSP matrix was separated into landrace (LR) and released cultivar (RC) sub-matrices, which showed significant differentiation in QTL-allele constitutions, with 58 LR alleles excluded and 16 new ones emerged in RC. Using the matrices, optimal crosses with great DT transgressive recombinants were predicted. From the detected QTL, 177 candidate genes were annotated and validated with quantitative Real-time PCR, and grouped into nine categories, with ABA and stress responders as the major parts. The key point of the above results is the establishment of relatively full QTL-allele matrices composed of numerous gene functions jointly conferring DT, therefore, demonstrates the complexity of DT genetic system and potential of CCSP in DT breeding.
干旱是影响植物生长和生产力的最重要因素之一。以前关于大豆耐旱性(DT)遗传系统的研究结果表明,不仅有少数几个基因参与了该性状,而且还涉及一个复杂的基因复合体。本研究的特点是,使用有效的限制两阶段多基因多等位基因全基因组关联研究,对两个综合的 DT 指标,相对植物重量(MPW)和高度(MPH)的隶属指数值,而不是单一的生物特征,对中国栽培大豆群体(CCSP)的一个大样本(564 个品系)进行了相对彻底的 QTL-等位基因/候选基因系统鉴定。基于 24694 个多等位基因标记,检测到与 MPW 和 MPH 相关的 75 个和 64 个 QTL,分别解释了 54.7%和 47.1%的表型方差。检测到的 QTL-等位基因被组织成每个指标的 QTL-等位基因矩阵,表明 DT 是由两个(甚至更多)亚性状的 QTL-等位基因系统赋予的超性状。每个 CCSP 矩阵被分为地方品种(LR)和已发布的栽培品种(RC)子矩阵,它们在 QTL-等位基因组成上表现出显著的差异,LR 中有 58 个等位基因被排除,RC 中有 16 个新等位基因出现。利用这些矩阵,可以预测具有巨大 DT 超越重组体的最佳杂交。从检测到的 QTL 中,注释和验证了 177 个候选基因,并分为 9 类,其中以 ABA 和应激反应为主。上述结果的关键点是建立了由众多基因功能共同赋予 DT 的相对完整的 QTL-等位基因矩阵,因此,展示了 DT 遗传系统的复杂性和 CCSP 在 DT 育种中的潜力。