Laboratory of Pharmacology and Brain Pathology, Humanitas Clinical and Research Center, Rozzano-Milan, Italy.
Department of Neuroscience, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milan, Italy.
PLoS Pathog. 2020 Jul 16;16(7):e1008654. doi: 10.1371/journal.ppat.1008654. eCollection 2020 Jul.
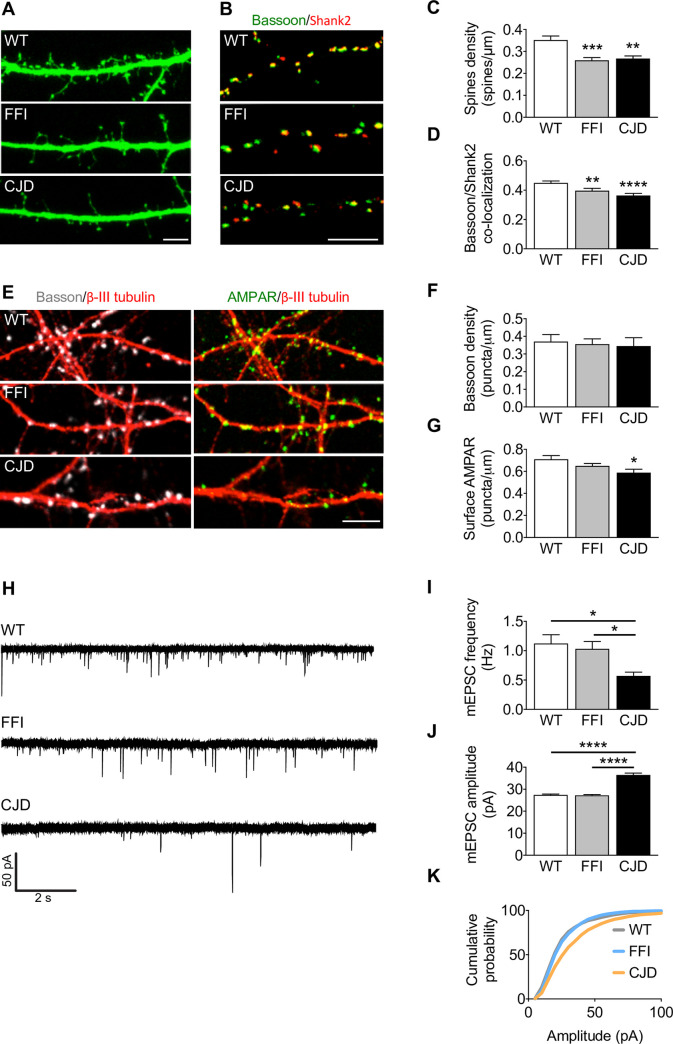
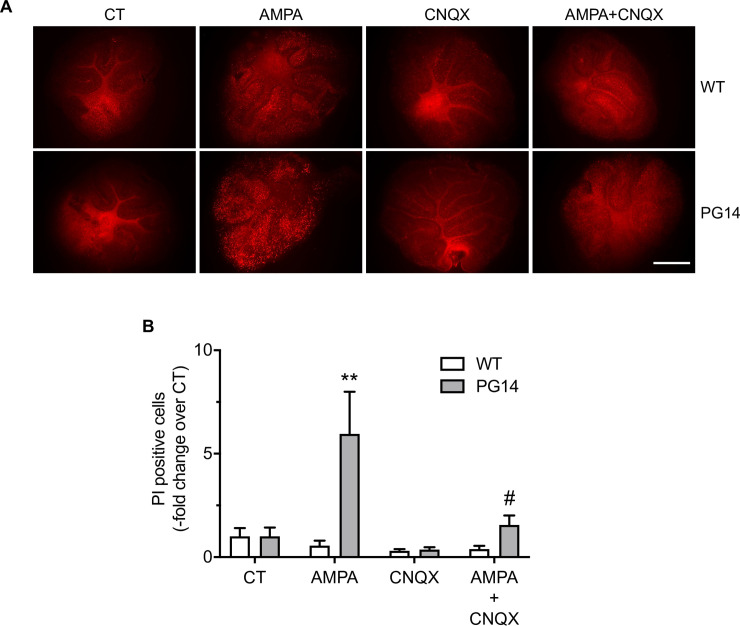
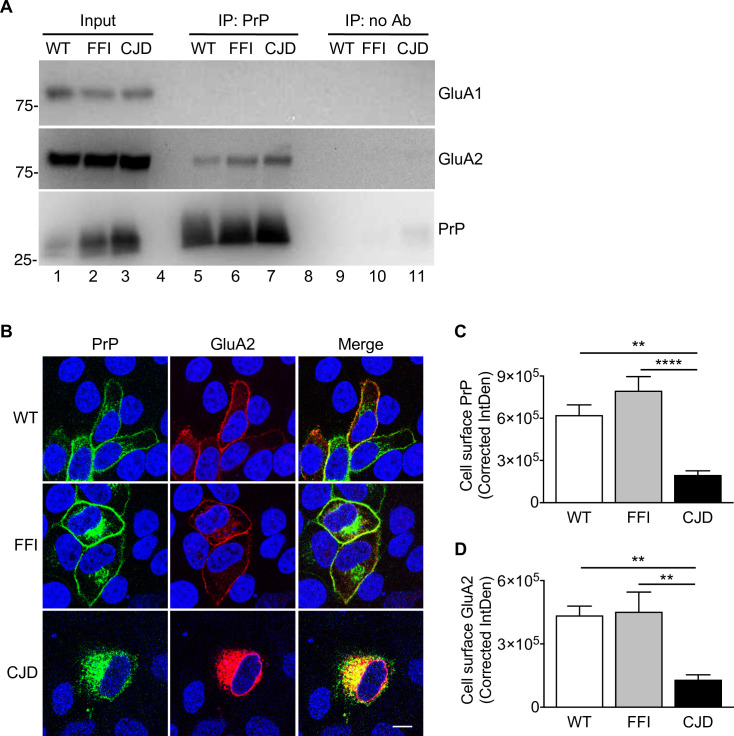
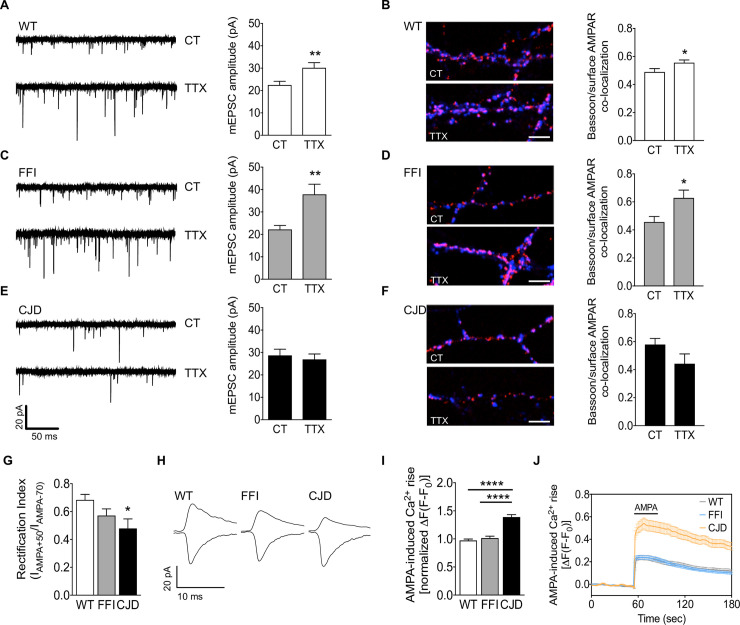
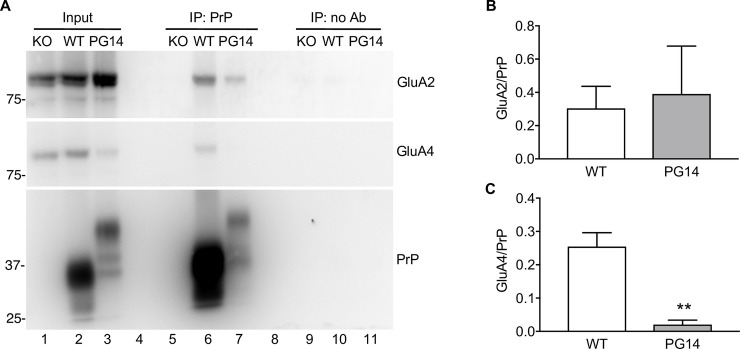
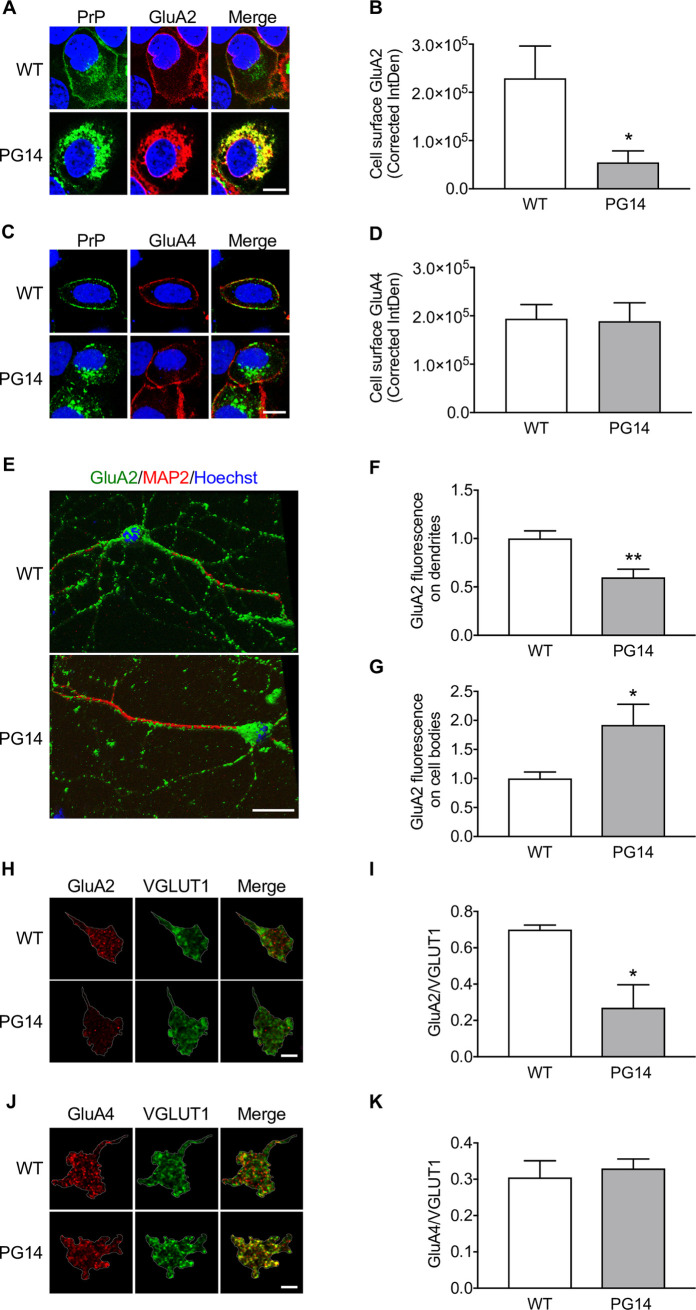
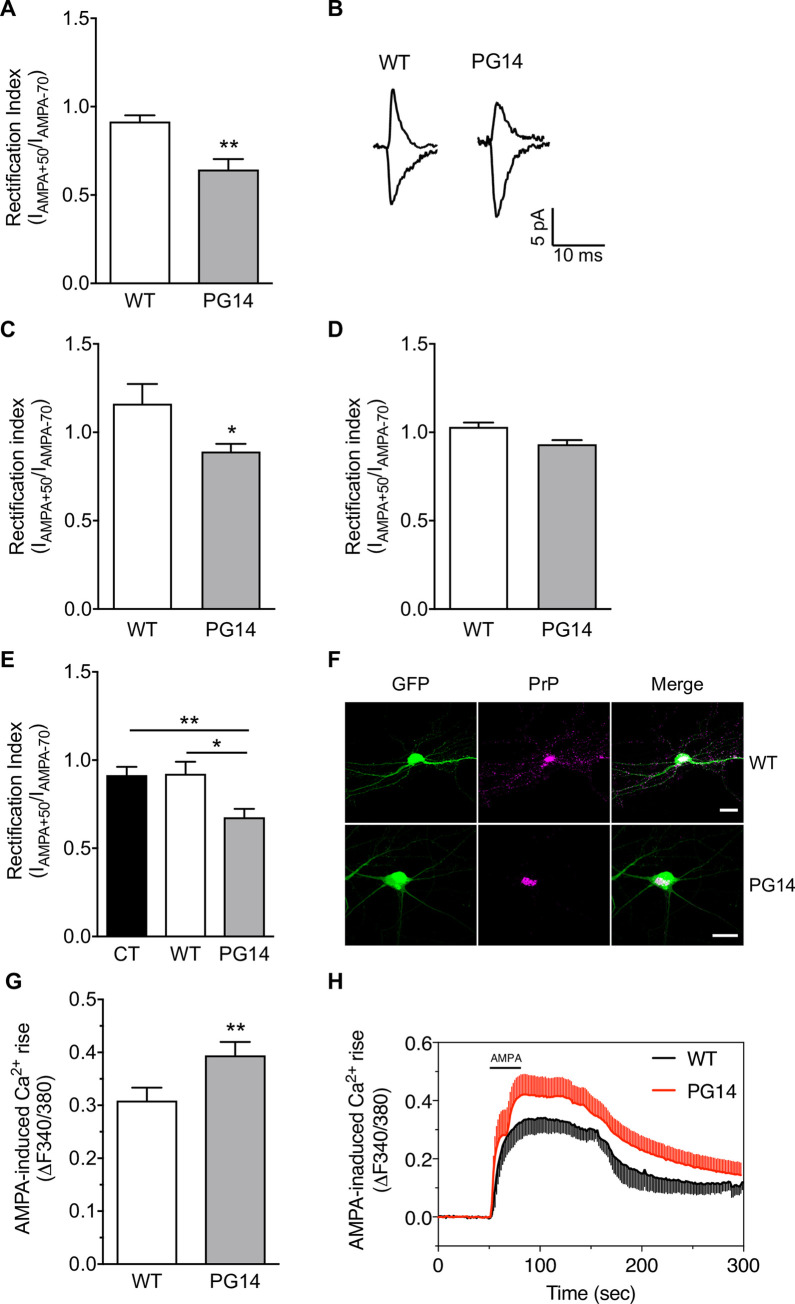
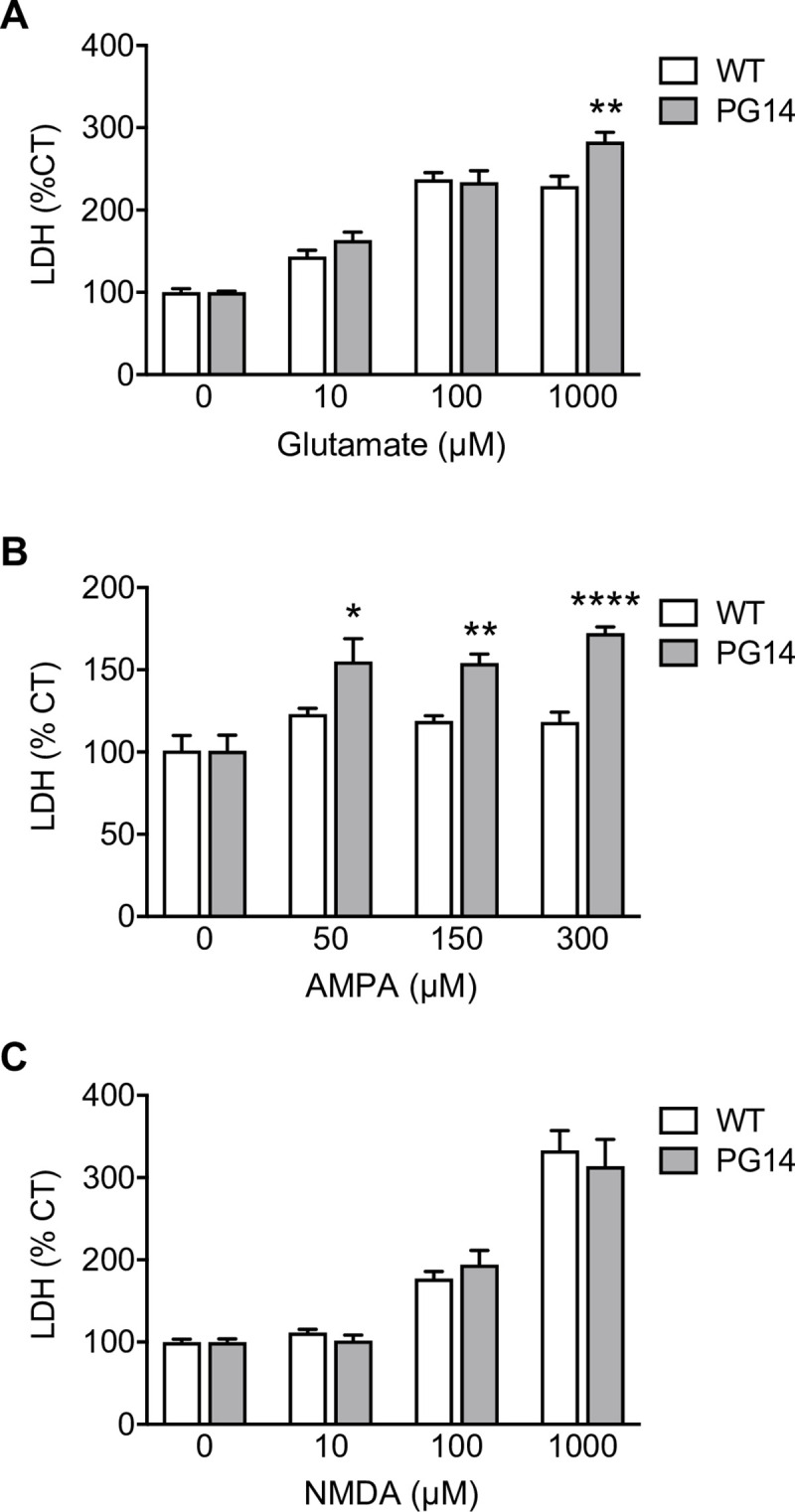
Prion protein (PrP) mutations are linked to genetic prion diseases, a class of phenotypically heterogeneous neurodegenerative disorders with invariably fatal outcome. How mutant PrP triggers neurodegeneration is not known. Synaptic dysfunction precedes neuronal loss but it is not clear whether, and through which mechanisms, disruption of synaptic activity ultimately leads to neuronal death. Here we show that mutant PrP impairs the secretory trafficking of AMPA receptors (AMPARs). Specifically, intracellular retention of the GluA2 subunit results in synaptic exposure of GluA2-lacking, calcium-permeable AMPARs, leading to increased calcium permeability and enhanced sensitivity to excitotoxic cell death. Mutant PrPs linked to different genetic prion diseases affect AMPAR trafficking and function in different ways. Our findings identify AMPARs as pathogenic targets in genetic prion diseases, and support the involvement of excitotoxicity in neurodegeneration. They also suggest a mechanistic explanation for how different mutant PrPs may cause distinct disease phenotypes.
朊病毒蛋白 (PrP) 突变与遗传朊病毒病有关,这是一类表型异质性的神经退行性疾病,具有不可避免的致命结局。突变型 PrP 如何引发神经退行性变尚不清楚。突触功能障碍先于神经元丢失,但尚不清楚突触活动的破坏是否以及通过哪些机制最终导致神经元死亡。在这里,我们表明突变型 PrP 会损害 AMPA 受体 (AMPAR) 的分泌性运输。具体而言,GluA2 亚基的细胞内滞留导致突触暴露于缺乏 GluA2、钙通透性的 AMPAR,导致钙通透性增加和对兴奋毒性细胞死亡的敏感性增强。与不同遗传朊病毒病相关的突变型 PrP 以不同的方式影响 AMPAR 的运输和功能。我们的发现将 AMPAR 确定为遗传朊病毒病的致病靶点,并支持兴奋性毒性在神经退行性变中的作用。它们还为不同的突变型 PrP 如何引起不同的疾病表型提供了一种机制解释。