Fralin Biomedical Research Institute at Virginia Tech Carilion, Roanoke, VA, USA.
Children's Hospital of Pittsburgh of UPMC, University of Pittsburgh, Pittsburgh, PA, USA.
Mol Genet Genomic Med. 2020 Oct;8(10):e1426. doi: 10.1002/mgg3.1426. Epub 2020 Jul 21.
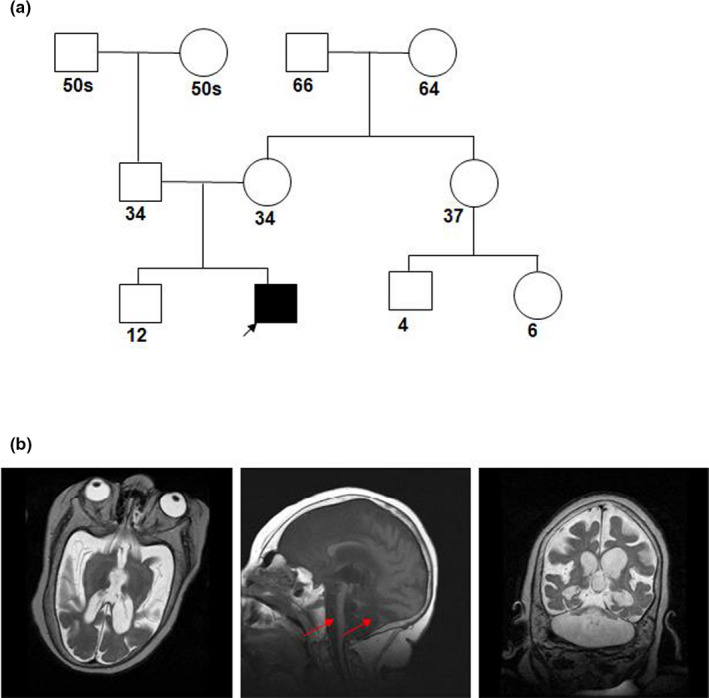
CASK is an X-linked gene in mammals and its deletion in males is incompatible with life. CASK heterozygous mutations in female patients associate with intellectual disability, microcephaly, pontocerebellar hypoplasia, and optic nerve hypoplasia, whereas CASK hemizygous mutations in males manifest as early infantile epileptic encephalopathy with a grim prognosis. Here, we report a rare case of survival of a male patient harboring a CASK null mutation to adolescent age.
Trio whole exome sequencing analysis was performed from blood genomic DNA. Magnetic resonance imaging (MRI), magnetic resonance spectroscopy (MRS), and electroencephalogram (EEG) analyses were performed to determine anomalies in brain development, metabolite concentrations, and electrical activity, respectively.
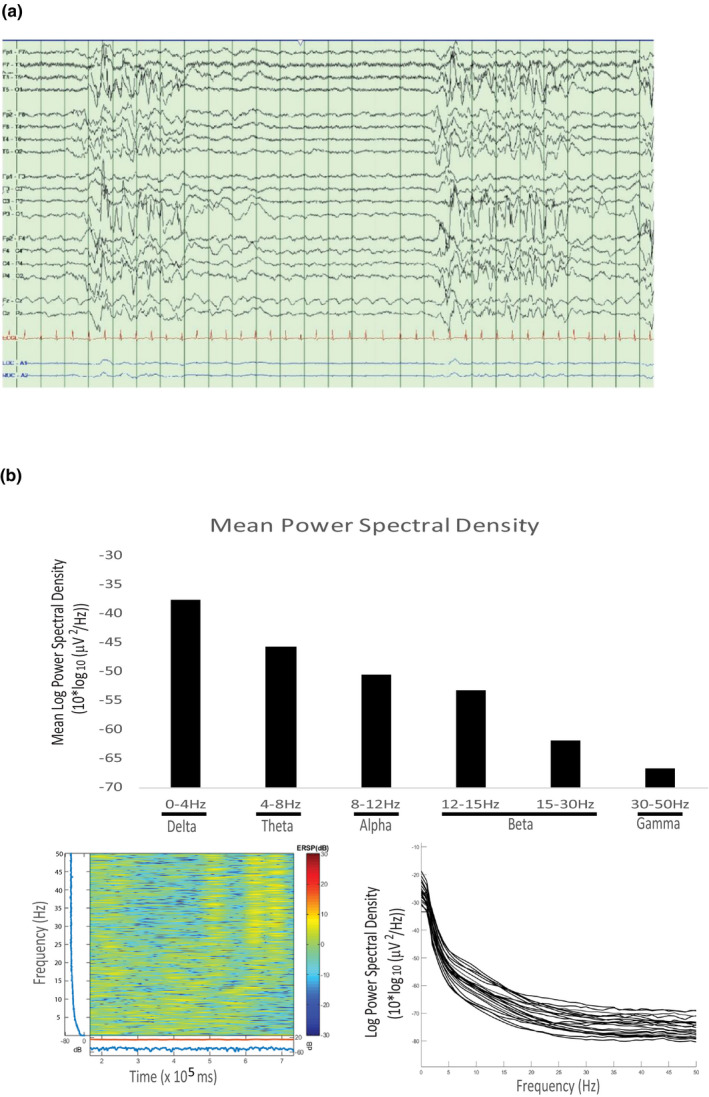
Trio-WES analysis identified a de novo c.79C>T (p.Arginine27Ter) mutation in CASK causing a premature translation termination at the very N-terminus of the protein. The 17-years, and 11-month-old male patient displayed profound intellectual disability, microcephaly, dysmorphism, ponto-cerebellar hypoplasia, and intractable epilepsy. His systemic symptoms included overall reduced somatic growth, dysautonomia, ventilator and G tube dependence, and severe osteopenia. Brain MRI revealed a severe cerebellar and brain stem hypoplasia with progressive cerebral atrophy. EEG spectral analysis revealed a global functional defect with generalized background slowing and delta waves dominating even in the awake state.
This case study is the first to report survival of a male patient carrying a CASK loss-of-function mutation to adolescence and highlights that improved palliative care could extend survival. Moreover, the genomic position encoding Arg27 in CASK may possess an increased susceptibility to mutations.
CASK 是哺乳动物的 X 连锁基因,其在男性中的缺失与生命不相容。女性患者的 CASK 杂合突变与智力残疾、小头畸形、桥脑小脑发育不良和视神经发育不良有关,而男性的 CASK 半合突变则表现为预后不良的早发性婴儿癫痫性脑病。在这里,我们报告了一例携带 CASK 无功能突变的男性患者存活至青少年期的罕见病例。
对来自血液基因组 DNA 的三核苷酸全外显子组测序分析。磁共振成像 (MRI)、磁共振波谱 (MRS) 和脑电图 (EEG) 分析分别用于确定脑发育、代谢物浓度和电活动的异常。
三核苷酸-WES 分析发现 CASK 中的一个新生 c.79C>T(p.Arg27Ter)突变,导致蛋白的非常 N 末端过早翻译终止。17 岁零 11 个月大的男性患者表现为严重的智力残疾、小头畸形、畸形、桥脑小脑发育不良和难治性癫痫。他的全身症状包括整体生长发育迟缓、自主神经功能障碍、呼吸机和 G 管依赖以及严重的骨质疏松症。脑部 MRI 显示严重的小脑和脑干发育不良伴进行性脑萎缩。脑电图频谱分析显示存在广泛的功能缺陷,表现为背景广泛减慢和 delta 波为主,即使在清醒状态下也是如此。
本病例研究首次报道了携带 CASK 功能丧失突变的男性患者存活至青少年期,并强调改善姑息治疗可以延长生存时间。此外,编码 CASK 中 Arg27 的基因组位置可能对突变具有更高的易感性。