Department of Systems Biology, City of Hope Comprehensive Cancer Center, Monrovia, CA, USA.
Dana Farber Cancer Institute, Boston, MA, USA.
Nature. 2020 Jul;583(7818):845-851. doi: 10.1038/s41586-020-2513-4. Epub 2020 Jul 22.
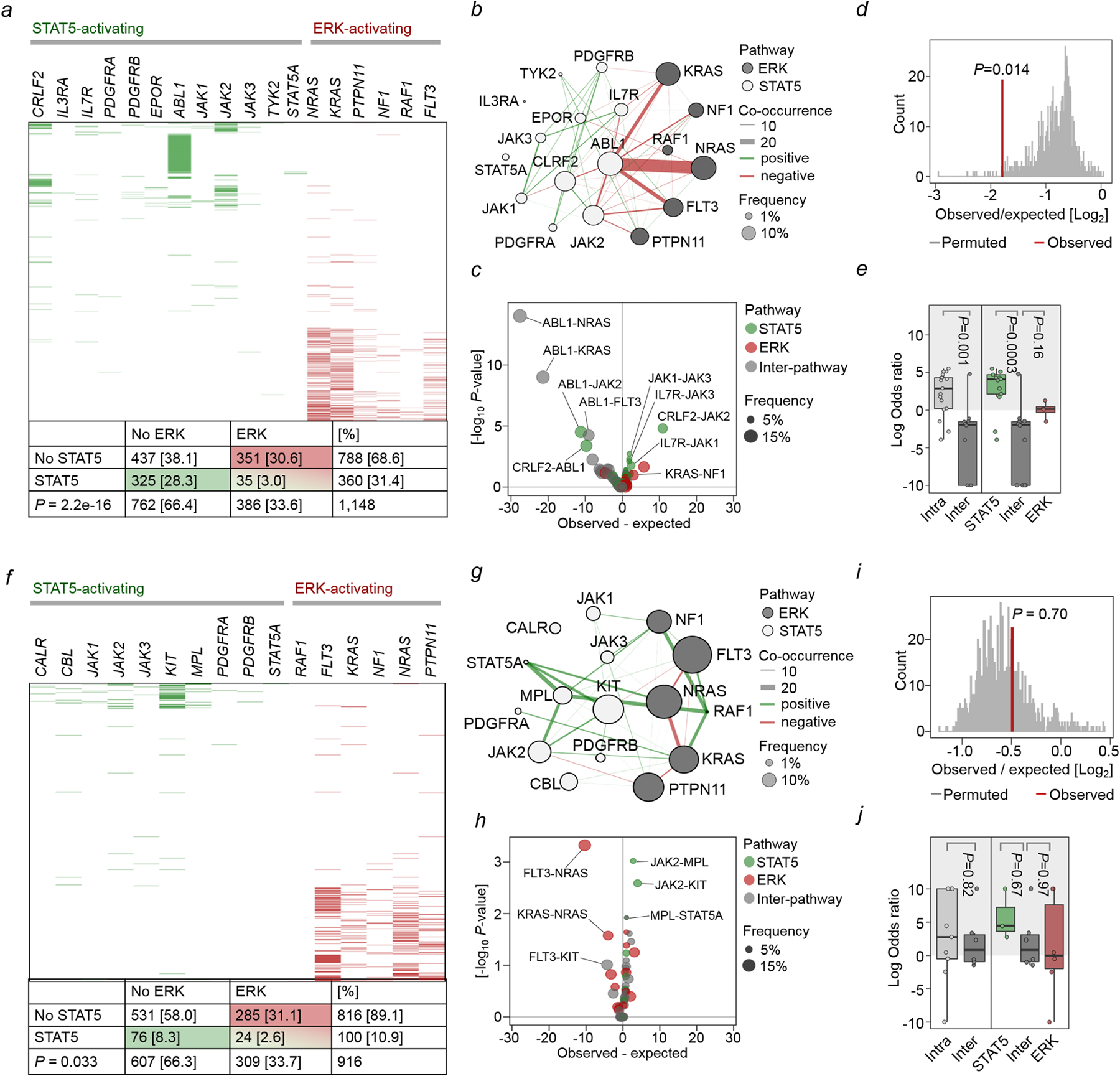
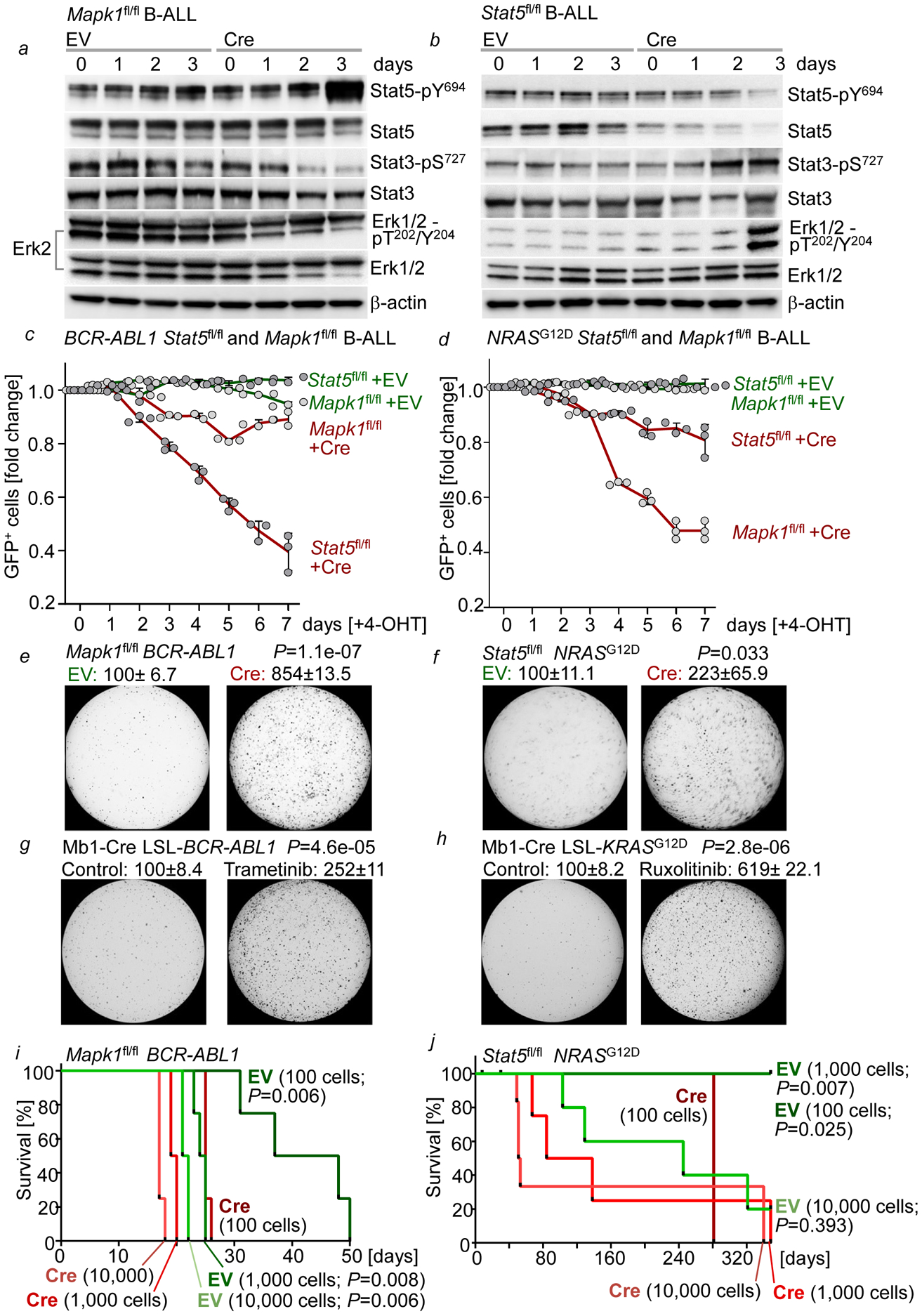
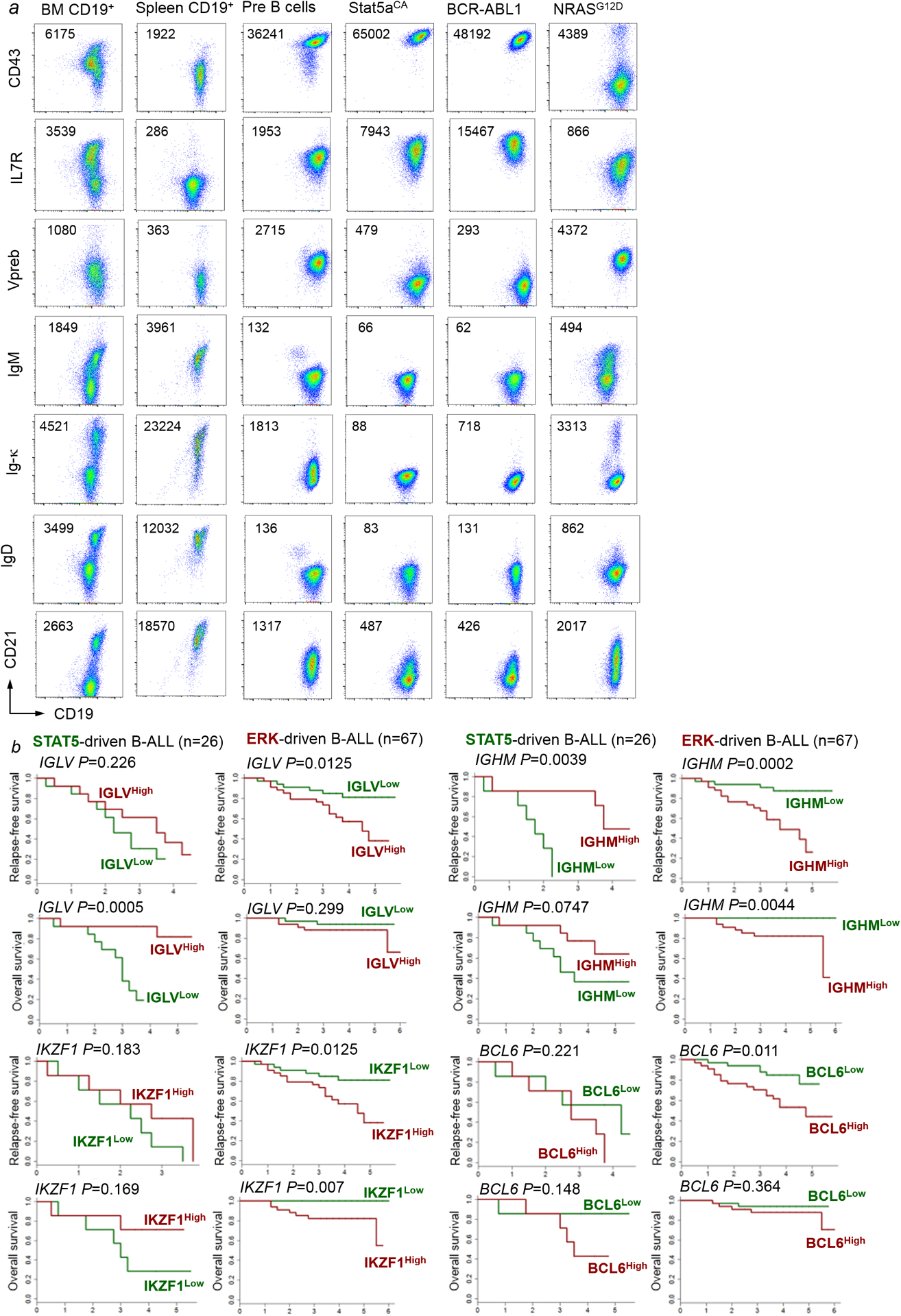
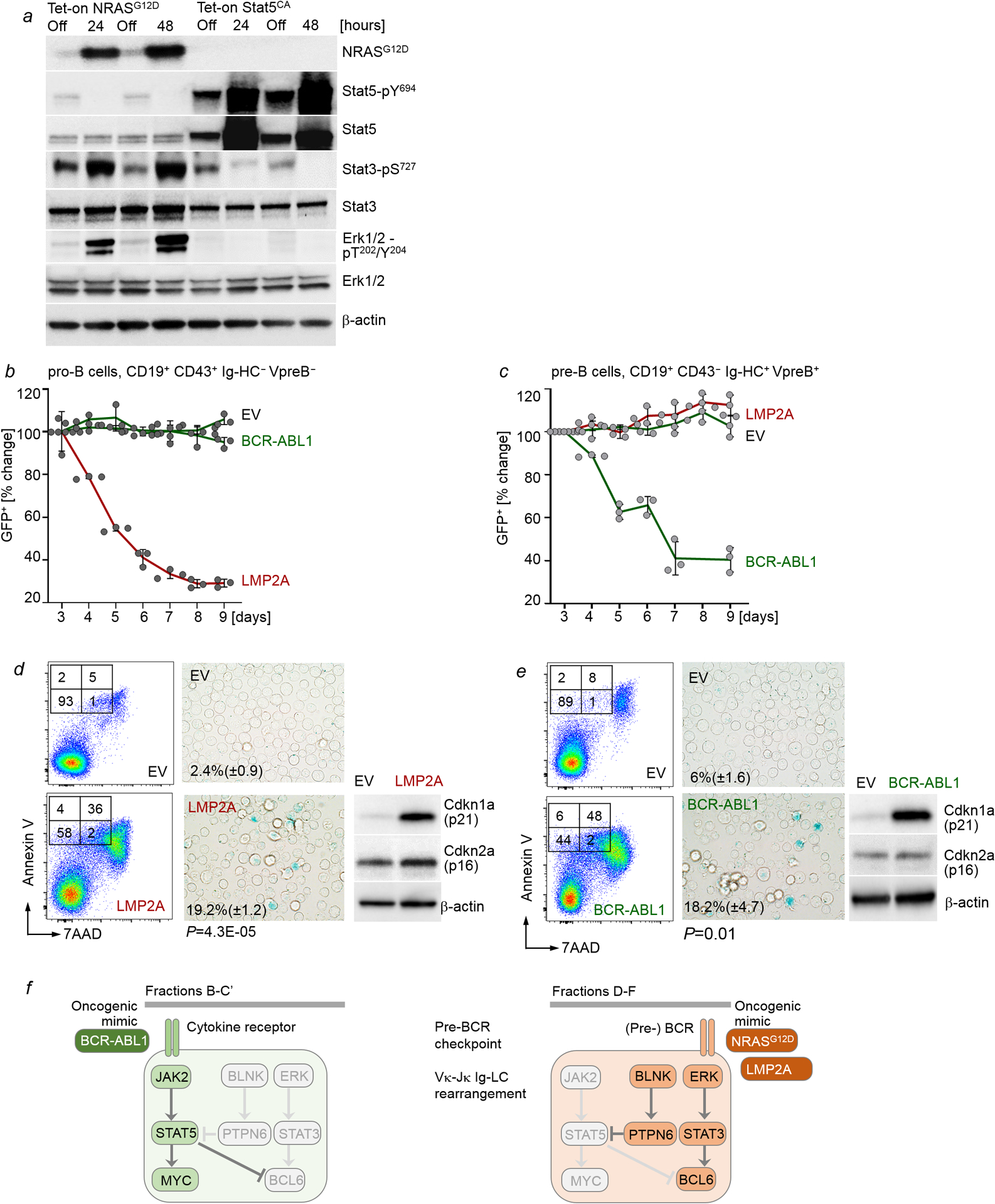
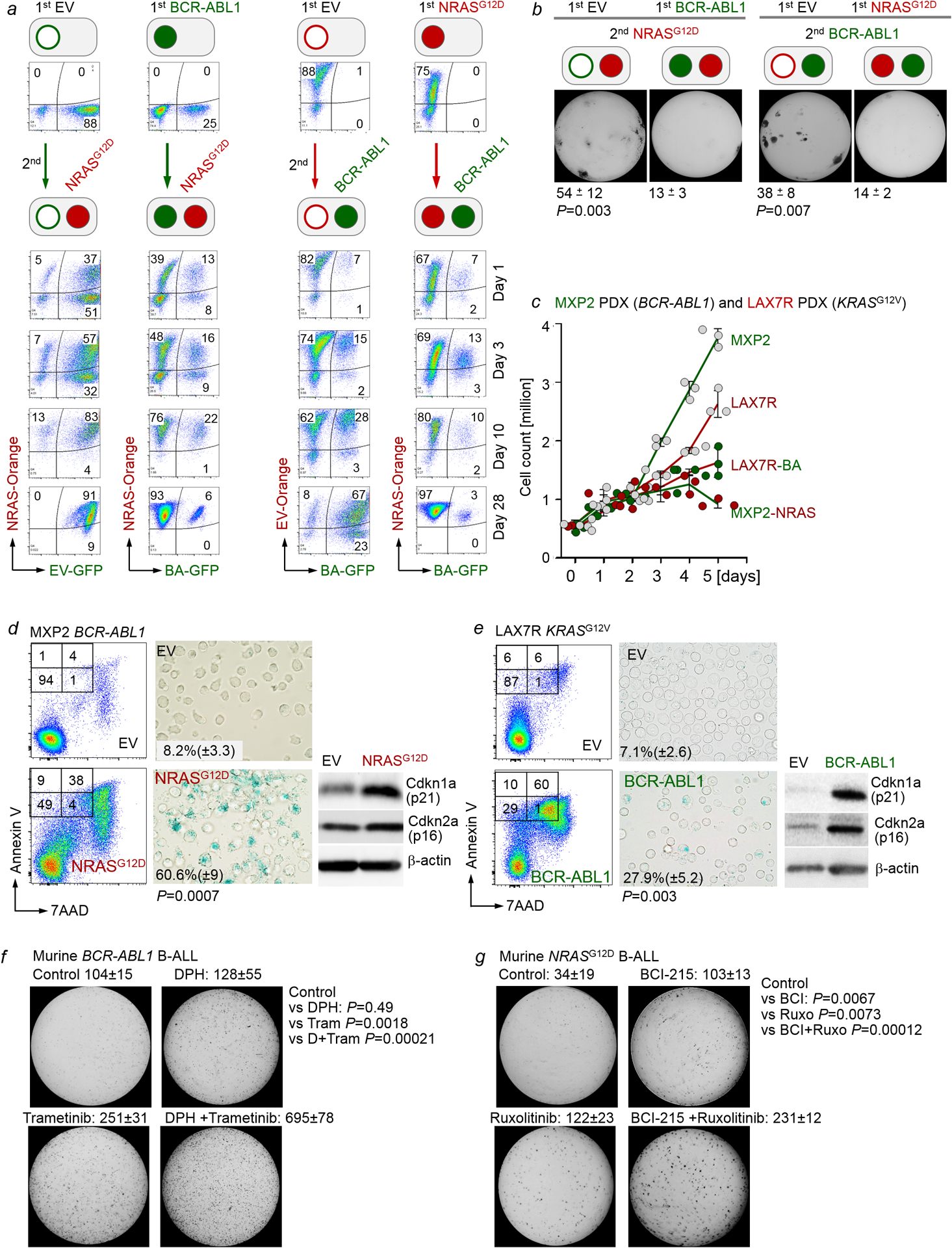
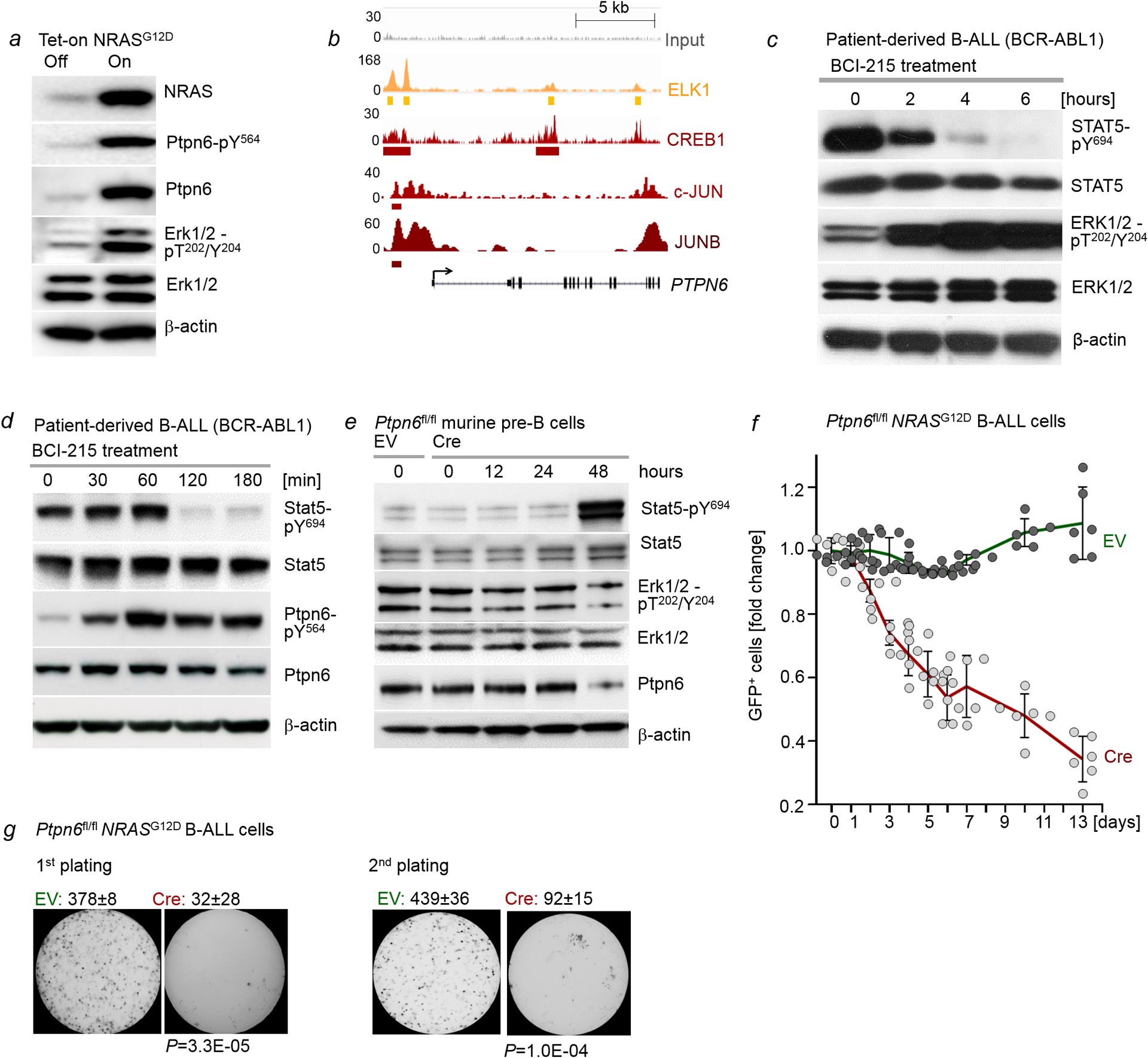
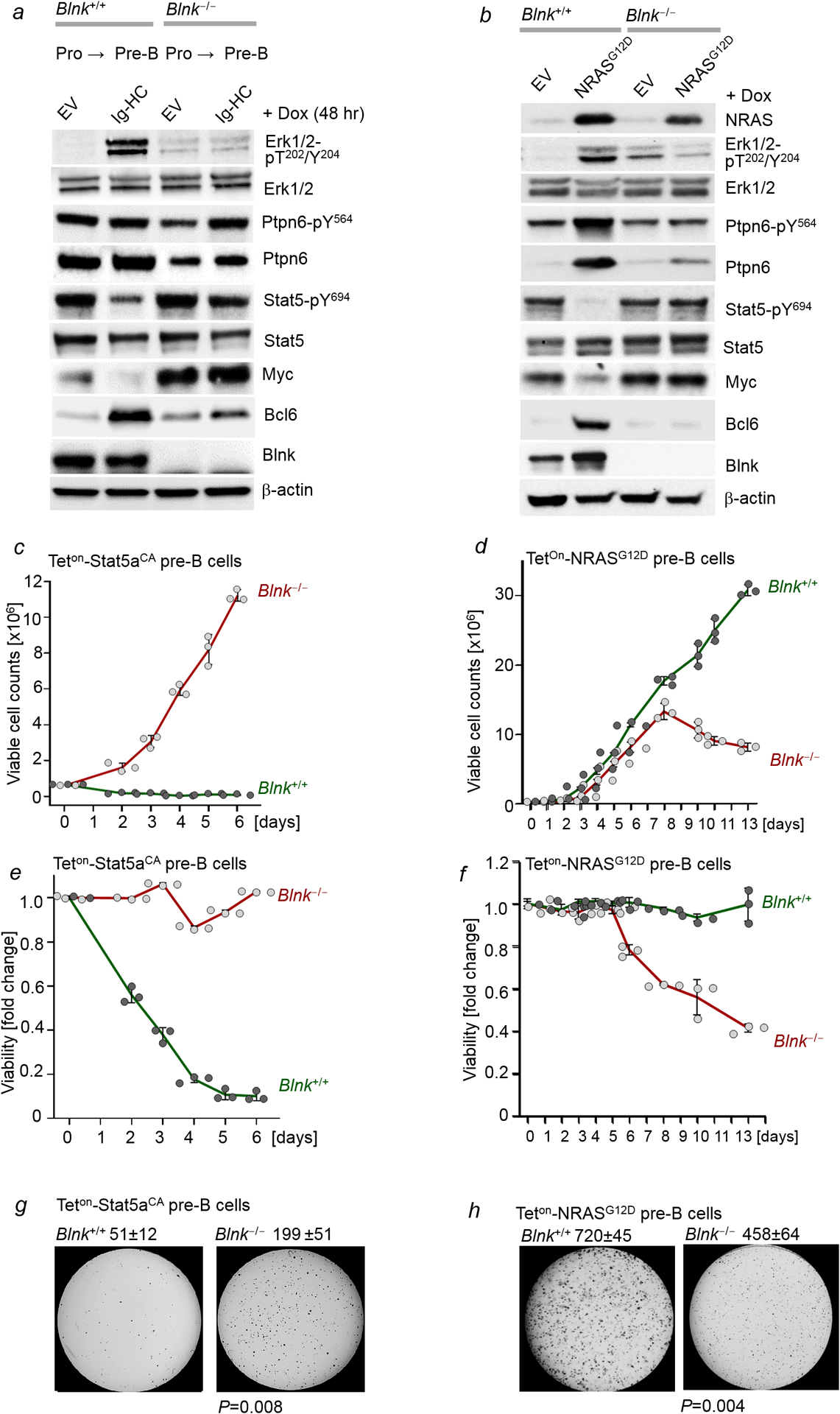
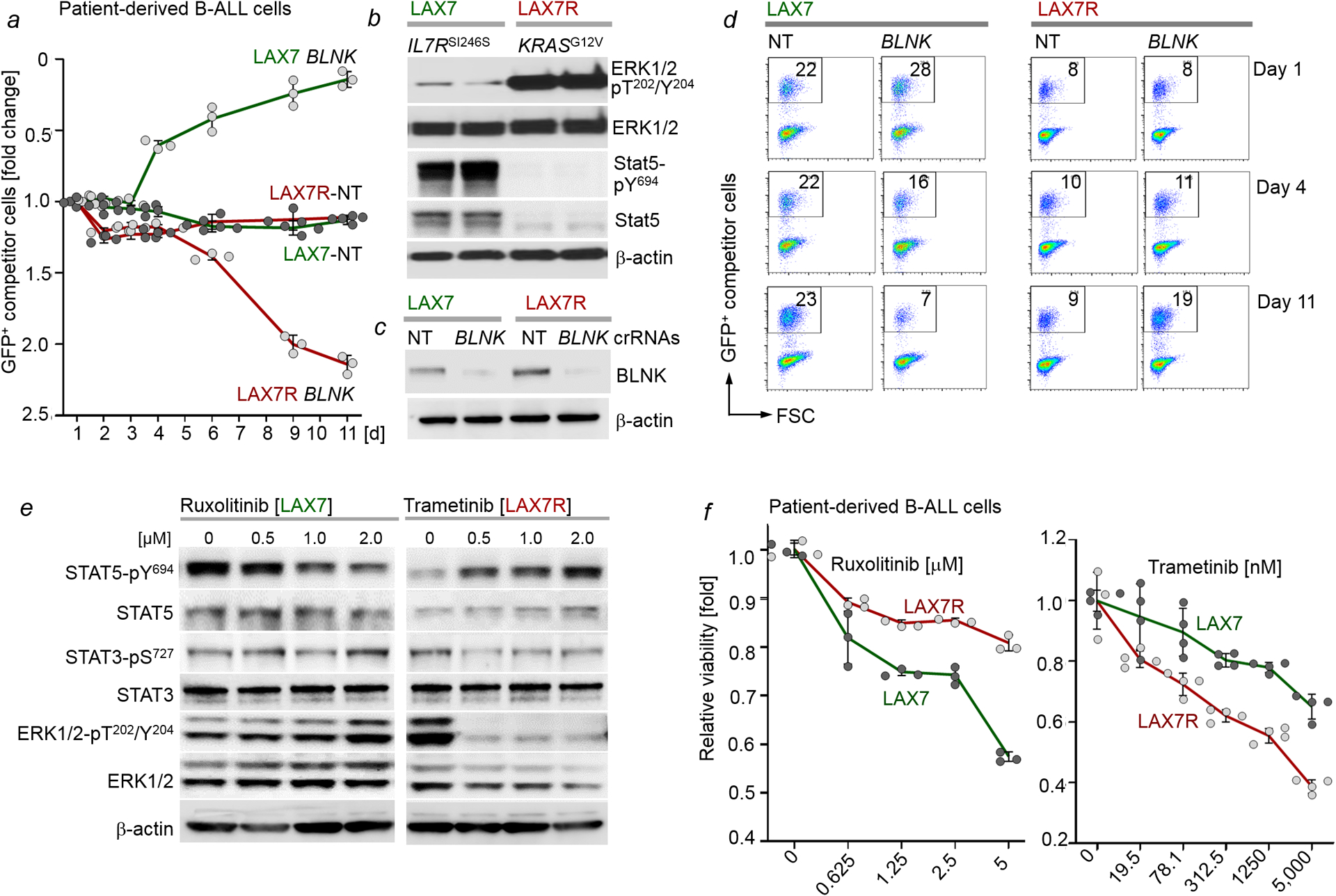
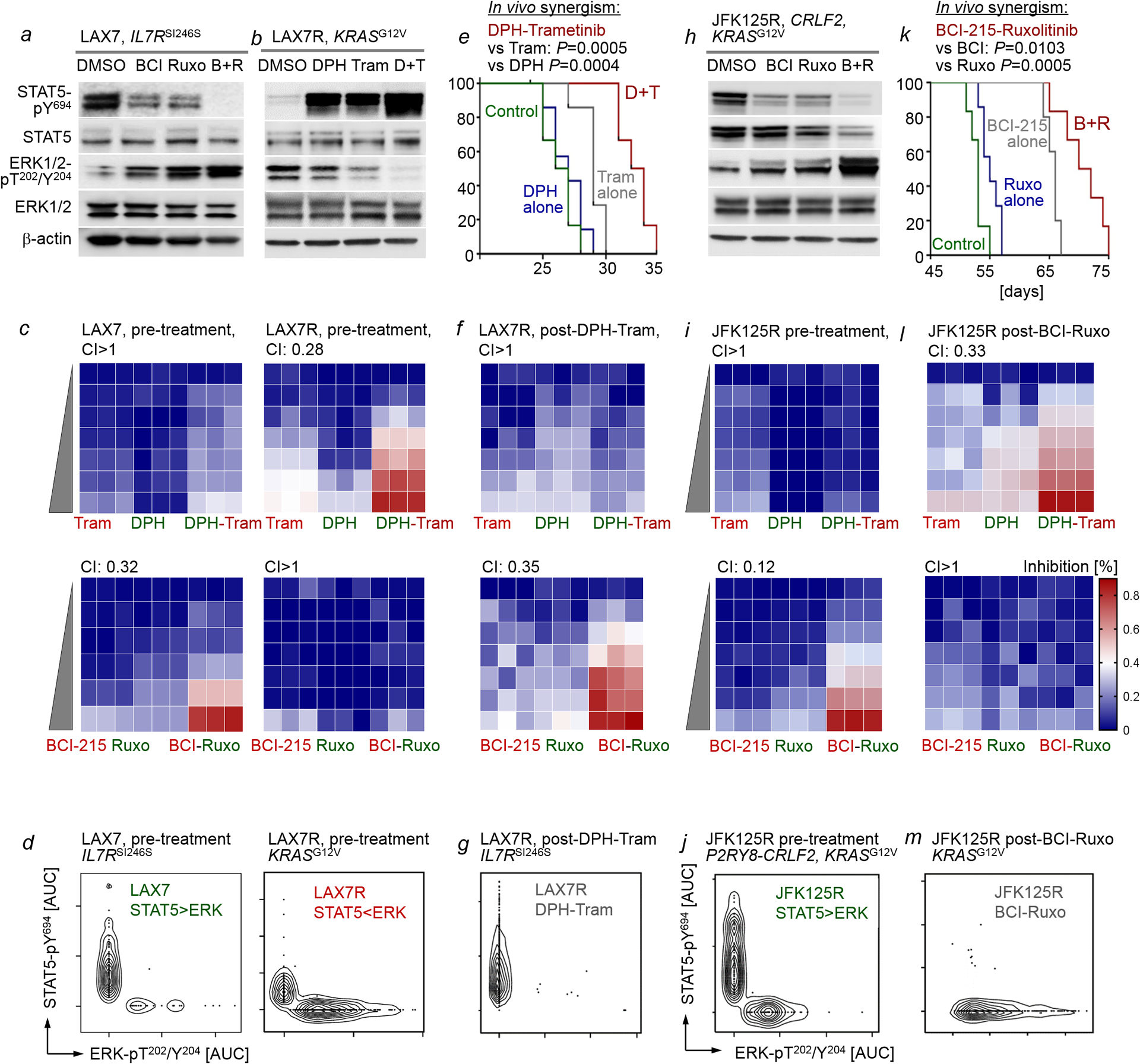
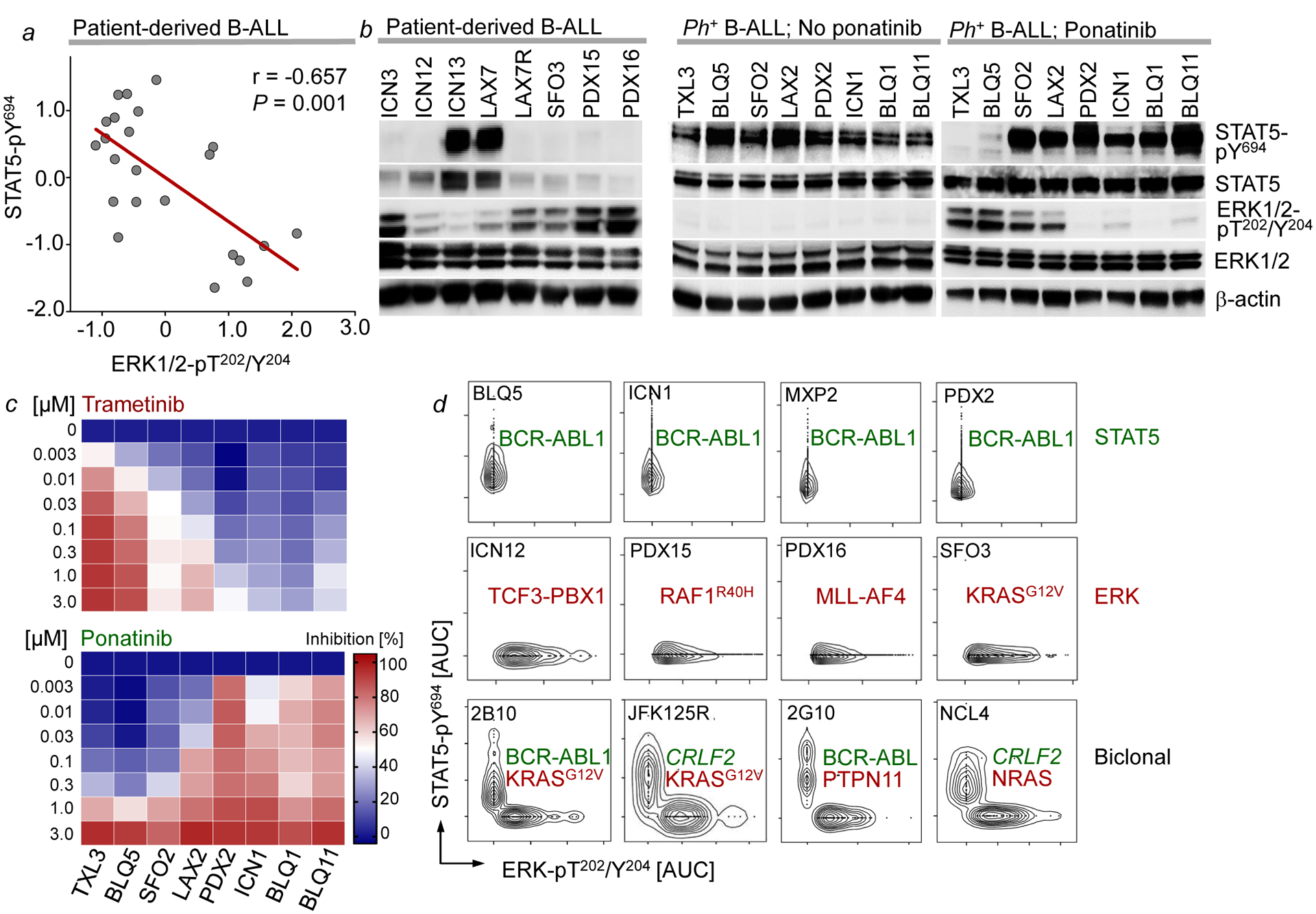
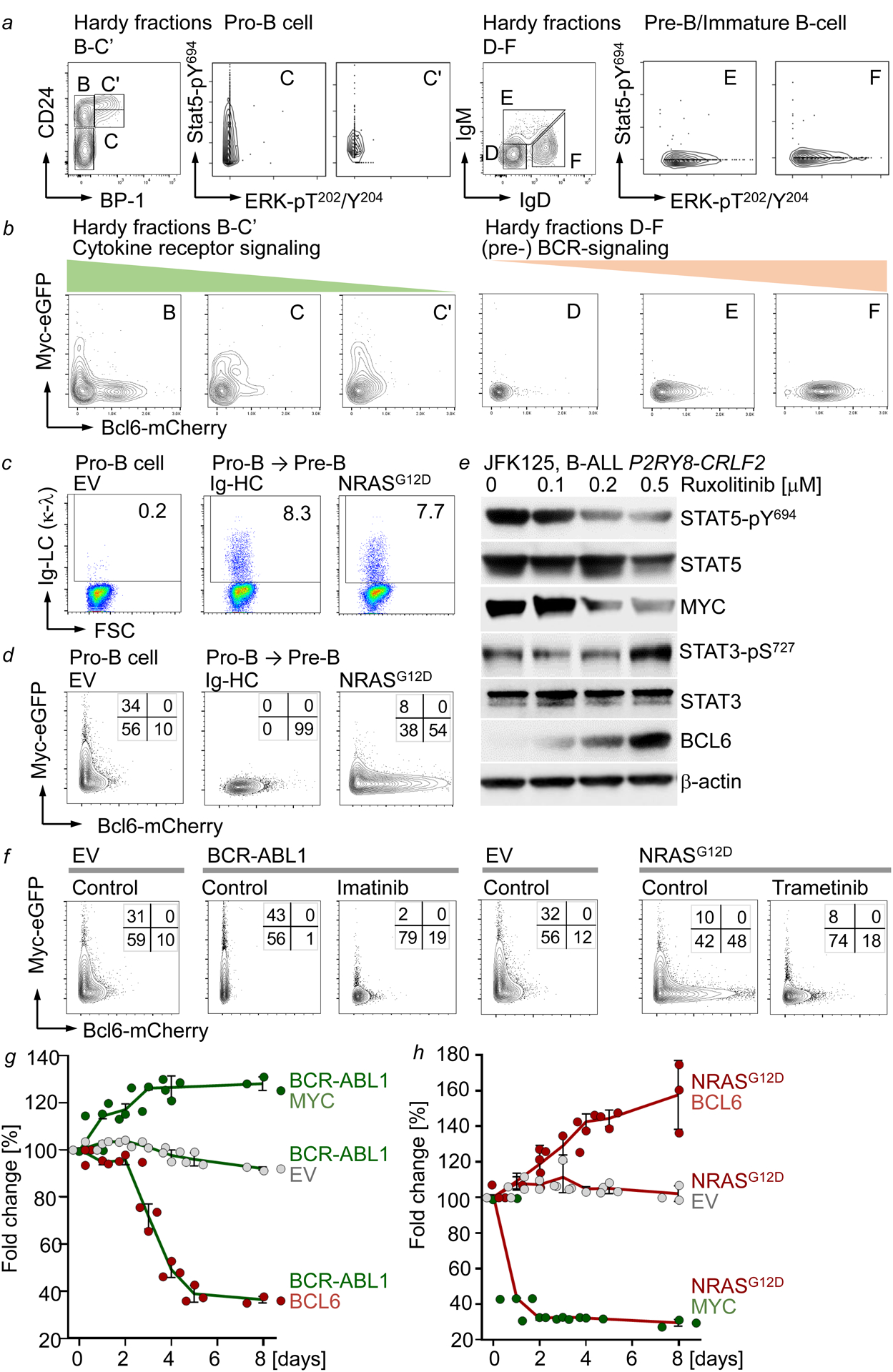
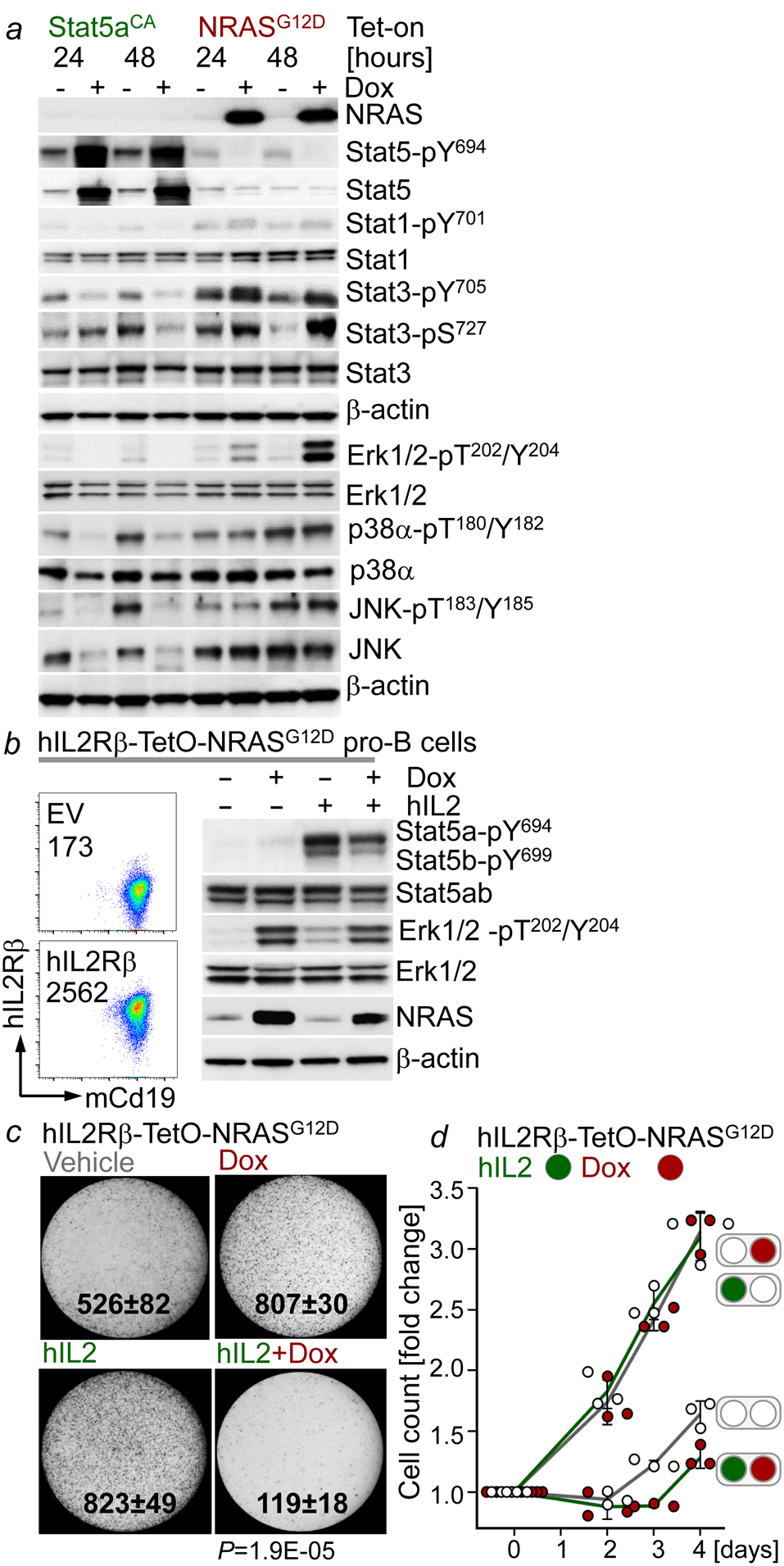
Malignant transformation of cells typically involves several genetic lesions, whose combined activity gives rise to cancer. Here we analyse 1,148 patient-derived B-cell leukaemia (B-ALL) samples, and find that individual mutations do not promote leukaemogenesis unless they converge on one single oncogenic pathway that is characteristic of the differentiation stage of transformed B cells. Mutations that are not aligned with this central oncogenic driver activate divergent pathways and subvert transformation. Oncogenic lesions in B-ALL frequently mimic signalling through cytokine receptors at the pro-B-cell stage (via activation of the signal-transduction protein STAT5) or pre-B-cell receptors in more mature cells (via activation of the protein kinase ERK). STAT5- and ERK-activating lesions are found frequently, but occur together in only around 3% of cases (P = 2.2 × 10). Single-cell mutation and phospho-protein analyses reveal the segregation of oncogenic STAT5 and ERK activation to competing clones. STAT5 and ERK engage opposing biochemical and transcriptional programs that are orchestrated by the transcription factors MYC and BCL6, respectively. Genetic reactivation of the divergent (suppressed) pathway comes at the expense of the principal oncogenic driver and reverses transformation. Conversely, deletion of divergent pathway components accelerates leukaemogenesis. Thus, persistence of divergent signalling pathways represents a powerful barrier to transformation, while convergence on one principal driver defines a central event in leukaemia initiation. Pharmacological reactivation of suppressed divergent circuits synergizes strongly with inhibition of the principal oncogenic driver. Hence, reactivation of divergent pathways can be leveraged as a previously unrecognized strategy to enhance treatment responses.
细胞的恶性转化通常涉及几种遗传损伤,其综合活性导致癌症的发生。在这里,我们分析了 1148 例患者来源的 B 细胞白血病(B-ALL)样本,发现单个突变不会促进白血病发生,除非它们集中在一个单一的致癌途径上,该途径是转化 B 细胞分化阶段的特征。与这个中央致癌驱动因素不一致的突变会激活不同的途径,并颠覆转化。B-ALL 中的致癌病变经常模拟在 pro-B 细胞阶段(通过激活信号转导蛋白 STAT5)或更成熟细胞中的 pre-B 细胞受体(通过激活蛋白激酶 ERK)的细胞因子受体的信号。STAT5 和 ERK 激活病变经常发生,但在大约 3%的病例中同时发生(P=2.2×10)。单细胞突变和磷酸蛋白分析揭示了致癌 STAT5 和 ERK 激活的分离到竞争克隆中。STAT5 和 ERK 参与对立的生化和转录程序,分别由转录因子 MYC 和 BCL6 协调。分歧(抑制)途径的遗传再激活以牺牲主要致癌驱动因素为代价并逆转转化。相反,分歧途径成分的缺失会加速白血病的发生。因此,分歧信号通路的持续存在代表了转化的强大障碍,而集中在一个主要驱动因素上定义了白血病起始的一个核心事件。抑制的分歧电路的药理学再激活与主要致癌驱动因素的抑制强烈协同。因此,重新激活分歧途径可以作为一种以前未被认识到的策略,以增强治疗反应。