Stanescu Sinziana, Belanger-Quintana Amaya, Alcalde Martin Carlos, Pérez-Cerdá Silvestre Celia, Merinero Cortés Begoña, Gonzalez Pérez Belen, Fernández García-Abril Carmen, Arrieta Blanco Francisco, Palacios Valverde Esperanza, Martínez-Pardo Casanova Mercedes
Unidad de Enfermedades Metabólicas, Hospital Ramón y Cajal, IRYCIS, Crta de Colmenar Viejo, Km 9,100, PC 28034, Madrid, Spain.
Servicio de Pediatria, Hospital Universitario Rio Hortega, C/Dulzaina 2, PC 47012, Valladolid, Spain.
Case Rep Pediatr. 2020 Jul 14;2020:1370293. doi: 10.1155/2020/1370293. eCollection 2020.
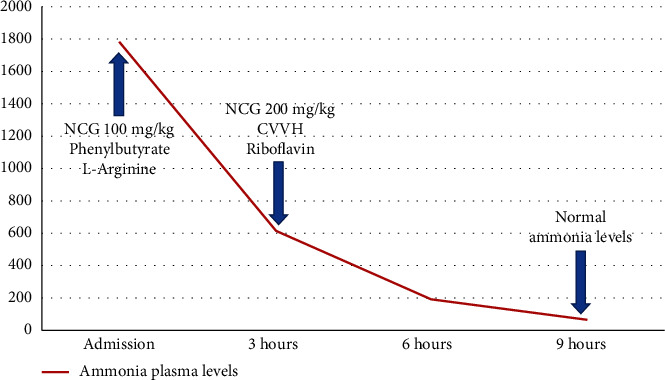
. Multiple acyl-CoA dehydrogenase deficiency is an autosomal recessive disorder of the amino acid metabolism and fatty acid oxidation due to the deficiency of the electron transfer protein or electron transfer protein ubiquinone oxidoreductase. The clinical picture ranges from a severe neonatal lethal presentation to late myopathic forms responsive to riboflavin. Up to now, there is no effective treatment for the neonatal form, which exhibits severe metabolic acidosis, hyperammonemia, hypoketotic hypoglycemia, and rhabdomyolysis. We present the case of a child who has had a good long-term outcome after a typical neonatal onset, with a dramatic drop in ammonia levels during the initial metabolic decompensation crisis and adequate control even during intercurrent diseases thereafter with N-carbamylglutamate treatment.
多种酰基辅酶A脱氢酶缺乏症是一种常染色体隐性氨基酸代谢和脂肪酸氧化障碍疾病,病因是电子传递蛋白或电子传递蛋白泛醌氧化还原酶缺乏。临床表现多样,从严重的新生儿致死型到对核黄素反应良好的晚期肌病型。到目前为止,新生儿型尚无有效治疗方法,其表现为严重代谢性酸中毒、高氨血症、低酮性低血糖和横纹肌溶解。我们报告一例典型新生儿起病的患儿,在最初的代谢失代偿危机期间氨水平急剧下降,此后即使在并发疾病期间经N-氨甲酰谷氨酸治疗也能得到充分控制,获得了良好的长期预后。