Robinson Katherine J, Watchon Maxinne, Laird Angela S
Centre for Motor Neuron Disease Research, Department of Biomedical Science, Faculty of Medicine, Health and Human Sciences, Macquarie University, Sydney, NSW, Australia.
Front Neurosci. 2020 Jul 16;14:707. doi: 10.3389/fnins.2020.00707. eCollection 2020.
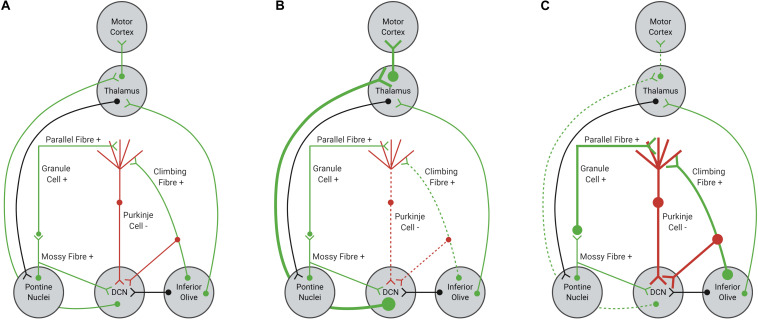
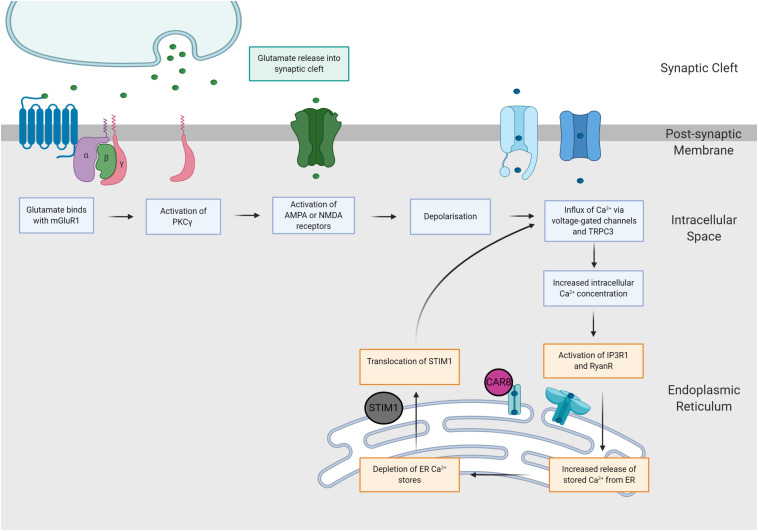
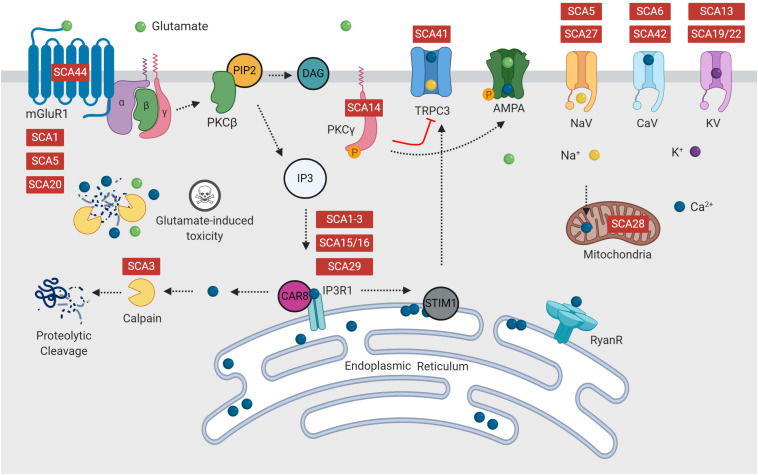
The spinocerebellar ataxias (SCAs) are a heterogeneous group of neurodegenerative diseases that share convergent disease features. A common symptom of these diseases is development of ataxia, involving impaired balance and motor coordination, usually stemming from cerebellar dysfunction and neurodegeneration. For most spinocerebellar ataxias, pathology can be attributed to an underlying gene mutation and the impaired function of the encoded protein through loss or gain-of-function effects. Strikingly, despite vast heterogeneity in the structure and function of disease-causing genes across the SCAs and the cellular processes affected, the downstream effects have considerable overlap, including alterations in cerebellar circuitry. Interestingly, aberrant function and degeneration of Purkinje cells, the major output neuronal population present within the cerebellum, precedes abnormalities in other neuronal populations within many SCAs, suggesting that Purkinje cells have increased vulnerability to cellular perturbations. Factors that are known to contribute to perturbed Purkinje cell function in spinocerebellar ataxias include altered gene expression resulting in altered expression or functionality of proteins and channels that modulate membrane potential, downstream impairments in intracellular calcium homeostasis and changes in glutamatergic input received from synapsing climbing or parallel fibers. This review will explore this enhanced vulnerability and the aberrant cerebellar circuitry linked with it in many forms of SCA. It is critical to understand why Purkinje cells are vulnerable to such insults and what overlapping pathogenic mechanisms are occurring across multiple SCAs, despite different underlying genetic mutations. Enhanced understanding of disease mechanisms will facilitate the development of treatments to prevent or slow progression of the underlying neurodegenerative processes, cerebellar atrophy and ataxic symptoms.
脊髓小脑共济失调(SCAs)是一组具有共同疾病特征的异质性神经退行性疾病。这些疾病的一个常见症状是共济失调的发展,包括平衡和运动协调受损,通常源于小脑功能障碍和神经退行性变。对于大多数脊髓小脑共济失调来说,病理可归因于潜在的基因突变以及编码蛋白通过功能丧失或功能获得效应而受损的功能。令人惊讶的是,尽管脊髓小脑共济失调中致病基因的结构和功能以及受影响的细胞过程存在巨大的异质性,但下游效应却有相当大的重叠,包括小脑回路的改变。有趣的是,浦肯野细胞(小脑内主要的输出神经元群体)的异常功能和退化在许多脊髓小脑共济失调中先于其他神经元群体出现异常,这表明浦肯野细胞对细胞扰动的易感性增加。已知在脊髓小脑共济失调中导致浦肯野细胞功能紊乱的因素包括基因表达改变,导致调节膜电位的蛋白质和通道的表达或功能改变、细胞内钙稳态的下游损伤以及从突触攀缘纤维或平行纤维接收的谷氨酸能输入的变化。本综述将探讨这种增强的易感性以及在多种形式的脊髓小脑共济失调中与之相关的异常小脑回路。了解为什么浦肯野细胞容易受到此类损伤以及尽管存在不同的潜在基因突变但在多种脊髓小脑共济失调中发生的重叠致病机制至关重要。对疾病机制的深入了解将有助于开发预防或减缓潜在神经退行性过程、小脑萎缩和共济失调症状进展的治疗方法。