Center of Reproduction, Development & Aging and Institute of Translation Medicine, Faculty of Health Sciences, University of Macau, Taipa, Macau, China.
Jiangsu Key Laboratory of Neuropsychiatric Diseases and College of Pharmaceutical Sciences, Soochow University, Suzhou, Jiangsu 215123, China.
Oxid Med Cell Longev. 2020 Jul 24;2020:2524174. doi: 10.1155/2020/2524174. eCollection 2020.
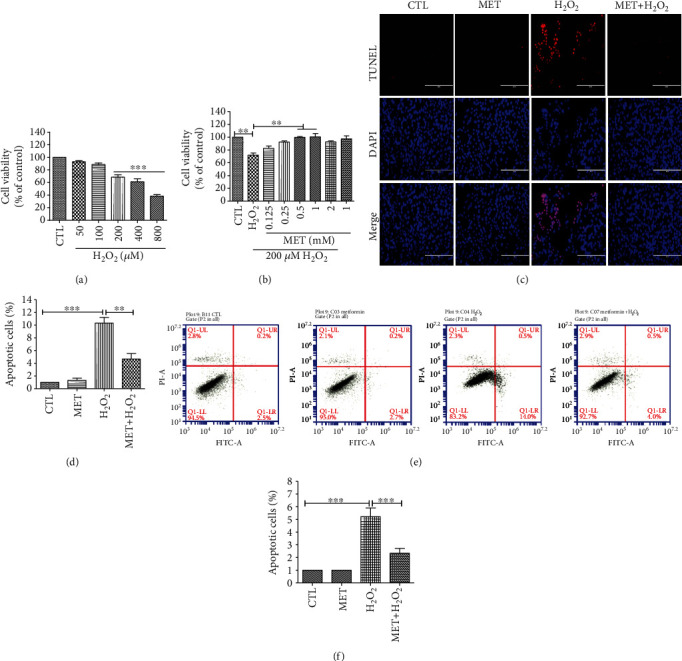
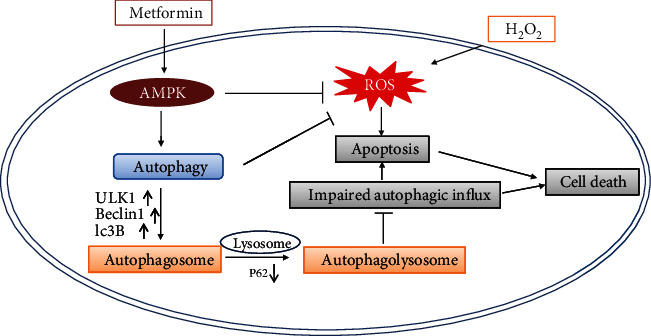
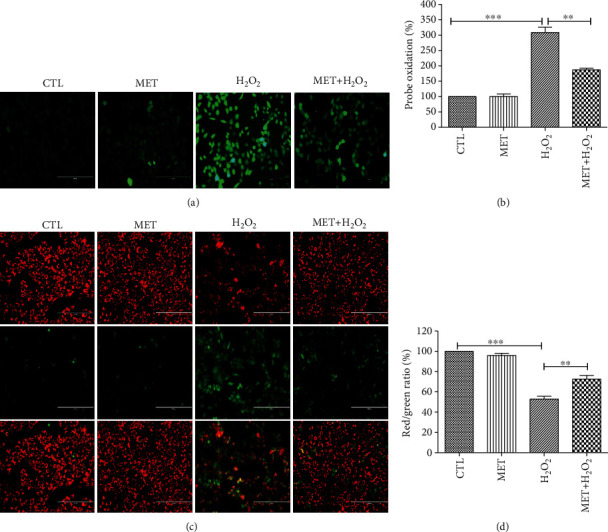
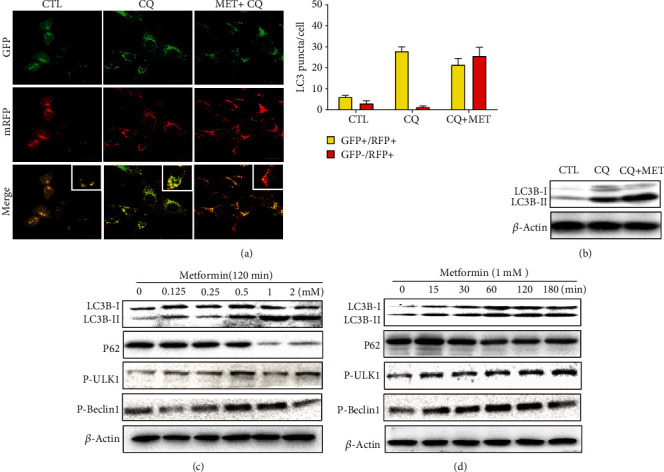
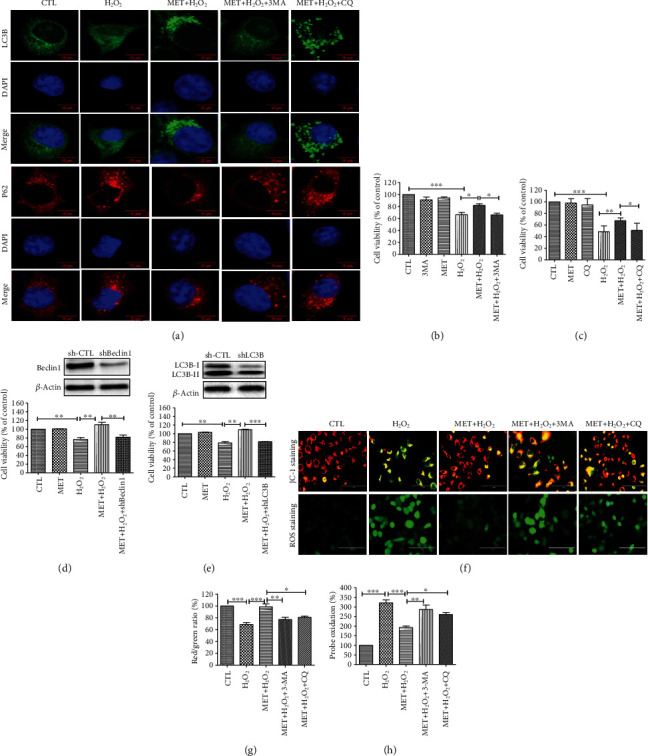
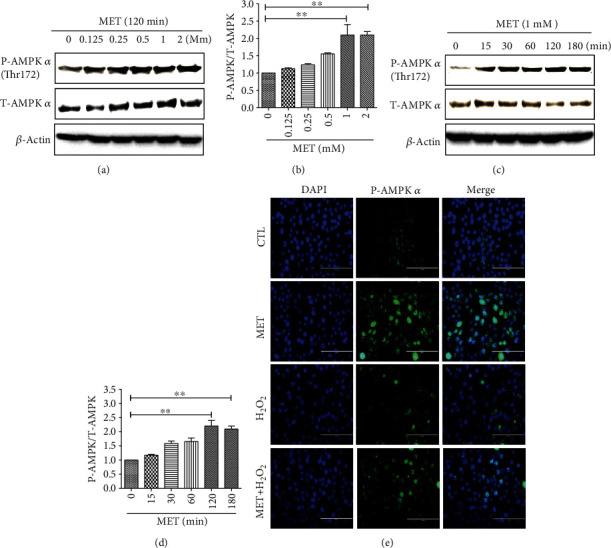
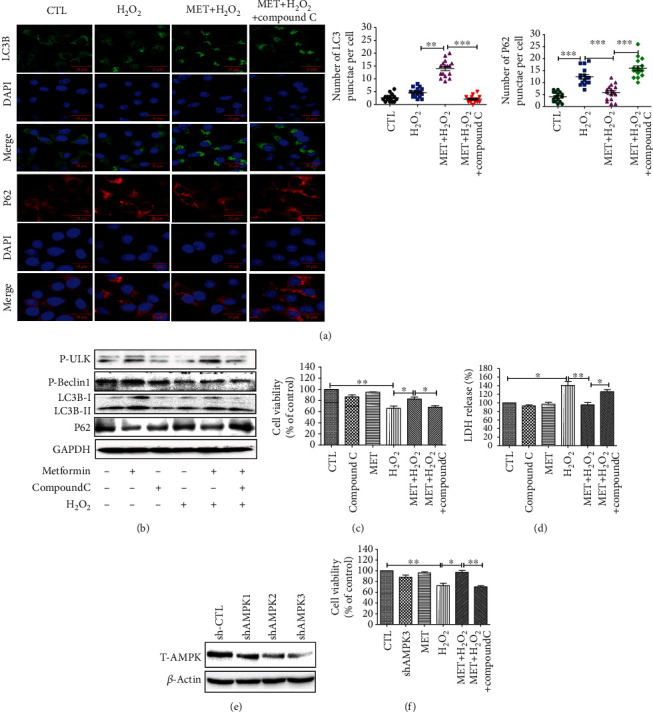
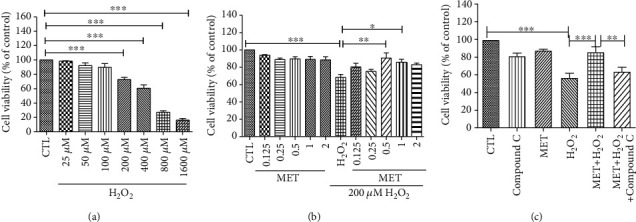
Age-related macular degeneration (AMD) is a leading cause of blindness with limited effective treatment. Although the pathogenesis of this disease is complex and not fully understood, the oxidative damage caused by excessive reactive oxygen species (ROS) in retinal pigment epithelium (RPE) has been considered as a major cause. Autophagy is essential for the degradation of cellular components damaged by ROS, and its dysregulation has been implicated in AMD pathogenesis. Therefore, strategies aiming to boost autophagy could be effective in protecting RPE cells from oxidative damage. Metformin is the first-line anti-type 2 diabetes drug and has been reported to stimulate autophagy in many tissues. We therefore hypothesized that metformin may be able to protect RPE cells against HO-induced oxidative damage by autophagy activation. In the present study, we found that metformin attenuated HO-induced cell viability loss, apoptosis, elevated ROS levels, and the collapse of the mitochondria membrane potential in D407 cells. Autophagy was stimulated by metformin, and inhibition of autophagy by 3-methyladenine (3-MA) and chloroquine (CQ) or knockdown of Beclin1 and LC3B blocked the protective effects of metformin. In addition, we showed that metformin could activate the AMPK pathway, whereas both pharmacological and genetic inhibitions of AMPK blocked the autophagy-stimulating and protective effects of metformin. Metformin conferred a similar protection against HO-induced oxidative damage in primary cultured human RPE cells. Taken together, these results demonstrate that metformin could protect RPE cells from HO-induced oxidative damage by stimulating autophagy via the activation of the AMPK pathway, supporting its potential use in the prevention and treatment of AMD.
年龄相关性黄斑变性(AMD)是一种导致失明的主要原因,目前治疗方法有限。虽然这种疾病的发病机制很复杂,尚未完全阐明,但视网膜色素上皮(RPE)中过量活性氧(ROS)引起的氧化损伤已被认为是主要原因之一。自噬对于降解由 ROS 引起的细胞成分至关重要,其失调与 AMD 的发病机制有关。因此,旨在增强自噬的策略可能有助于保护 RPE 细胞免受氧化损伤。二甲双胍是治疗 2 型糖尿病的一线药物,据报道它能在许多组织中刺激自噬。因此,我们假设二甲双胍可能通过激活自噬来保护 RPE 细胞免受 HO 诱导的氧化损伤。在本研究中,我们发现二甲双胍能减轻 HO 诱导的 D407 细胞活力丧失、凋亡、ROS 水平升高和线粒体膜电位崩溃。二甲双胍能刺激自噬,而 3-甲基腺嘌呤(3-MA)和氯喹(CQ)抑制自噬或敲低 Beclin1 和 LC3B 则阻断了二甲双胍的保护作用。此外,我们表明二甲双胍可以激活 AMPK 通路,而 AMPK 的药理学和遗传学抑制则阻断了二甲双胍的自噬刺激和保护作用。二甲双胍对原代培养的人 RPE 细胞中 HO 诱导的氧化损伤也有类似的保护作用。综上所述,这些结果表明,二甲双胍通过激活 AMPK 通路刺激自噬来保护 RPE 细胞免受 HO 诱导的氧化损伤,这支持了它在 AMD 的预防和治疗中的潜在用途。