Robak Laurie A, Du Renqian, Yuan Bo, Gu Shen, Alfradique-Dunham Isabel, Kondapalli Vismaya, Hinojosa Evelyn, Stillwell Amanda, Young Emily, Zhang Chaofan, Song Xiaofei, Du Haowei, Gambin Tomasz, Jhangiani Shalini N, Coban Akdemir Zeynep, Muzny Donna M, Tejomurtula Anusha, Ross Owen A, Shaw Chad, Jankovic Joseph, Bi Weimin, Posey Jennifer E, Lupski James R, Shulman Joshua M
Department of Molecular and Human Genetics (L.A.R., R.D., B.Y., S.G., V.K., E.H., A.S., E.Y., C.Z., X.S., H.D., T.G., Z.C.A., A.T., C.S., W.B., J.E.P., J.R.L., J.M.S.), Department of Neurology (I.A.-D., J.J., J.M.S.), and Human Genome Sequencing Center (S.N.J., D.M.M., J.R.L.), Baylor College of Medicine, Houston, TX; Baylor Genetics (W.B.), Houston, TX; Department of Neurology (O.A.R.), Department of Neuroscience (O.A.R.), and Department of Clinical Genomics (O.A.R.), Mayo Clinic, Jacksonville, FL; Parkinson's Disease Center and Movement Disorders Clinic (J.J.) and Department of Pediatrics (J.R.L., J.M.S.), Baylor College of Medicine, Houston, TX; Department of Pediatrics (J.R.L.), Texas Children's Hospital, Houston; Department of Neuroscience (J.M.S.), Baylor College of Medicine, Houston, TX; and Jan and Dan Duncan Neurological Research Institute (J.M.S.), Texas Children's Hospital, Houston.
Neurol Genet. 2020 Jul 28;6(5):e498. doi: 10.1212/NXG.0000000000000498. eCollection 2020 Oct.
To determine how single nucleotide variants (SNVs) and copy number variants (CNVs) contribute to molecular diagnosis in familial Parkinson disease (PD), we integrated exome sequencing (ES) and genome-wide array-based comparative genomic hybridization (aCGH) and further probed CNV structure to reveal mutational mechanisms.
We performed ES on 110 subjects with PD and a positive family history; 99 subjects were also evaluated using genome-wide aCGH. We interrogated ES and aCGH data for pathogenic SNVs and CNVs at Mendelian PD gene loci. We confirmed SNVs via Sanger sequencing and further characterized CNVs with custom-designed high-density aCGH, droplet digital PCR, and breakpoint sequencing.
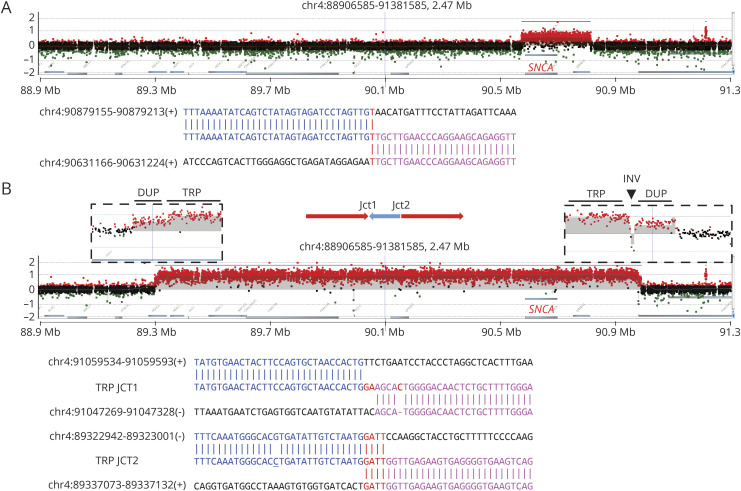
Using ES, we discovered individuals with known pathogenic SNVs in (p.Glu365Lys, p.Thr408Met, p.Asn409Ser, and p.Leu483Pro) and (p.Arg1441Gly and p.Gly2019Ser). Two subjects were each double heterozygotes for variants in and . Based on aCGH, we additionally discovered cases with an duplication and heterozygous intragenic deletion. Five additional subjects harbored both SNVs (p.Asn52Metfs29, p.Thr240Met, p.Pro437Leu, and p.Trp453) and likely disrupting CNVs at the locus, consistent with compound heterozygosity. In nearly all cases, breakpoint sequencing revealed microhomology, a mutational signature consistent with CNV formation due to DNA replication errors.
Integrated ES and aCGH yielded a genetic diagnosis in 19.3% of our familial PD cohort. Our analyses highlight potential mechanisms for and CNV formation, uncover multilocus pathogenic variation, and identify novel SNVs and CNVs for further investigation as potential PD risk alleles.
为了确定单核苷酸变异(SNV)和拷贝数变异(CNV)如何有助于家族性帕金森病(PD)的分子诊断,我们整合了外显子组测序(ES)和基于全基因组阵列的比较基因组杂交(aCGH),并进一步探究CNV结构以揭示突变机制。
我们对110名有PD且家族史呈阳性的受试者进行了ES;99名受试者还使用全基因组aCGH进行了评估。我们在孟德尔PD基因位点询问ES和aCGH数据以查找致病性SNV和CNV。我们通过桑格测序确认SNV,并使用定制设计的高密度aCGH、液滴数字PCR和断点测序进一步表征CNV。
使用ES,我们在(p.Glu365Lys、p.Thr408Met、p.Asn409Ser和p.Leu483Pro)和(p.Arg1441Gly和p.Gly2019Ser)中发现了具有已知致病性SNV的个体。两名受试者分别是和变异的双杂合子。基于aCGH,我们还发现了有重复和基因内杂合缺失的病例。另外五名受试者同时携带SNV(p.Asn52Metfs29、p.Thr240Met、p.Pro437Leu和p.Trp453)以及可能破坏基因座处CNV,这与复合杂合性一致。在几乎所有病例中,断点测序都揭示了微同源性,这是一种与DNA复制错误导致的CNV形成一致的突变特征。
整合ES和aCGH在我们19.3%的家族性PD队列中实现了基因诊断。我们的分析突出了和CNV形成的潜在机制,揭示了多位点致病性变异,并鉴定了新的SNV和CNV以供进一步研究作为潜在的PD风险等位基因。