Life Sciences & Health, Centrum Wiskunde & Informatica, Amsterdam 1098 XG, The Netherlands.
Theoretical Biology & Bioinformatics, Utrecht University, Utrecht 3512 JE, The Netherlands.
Bioinformatics. 2021 May 17;37(7):905-912. doi: 10.1093/bioinformatics/btaa760.
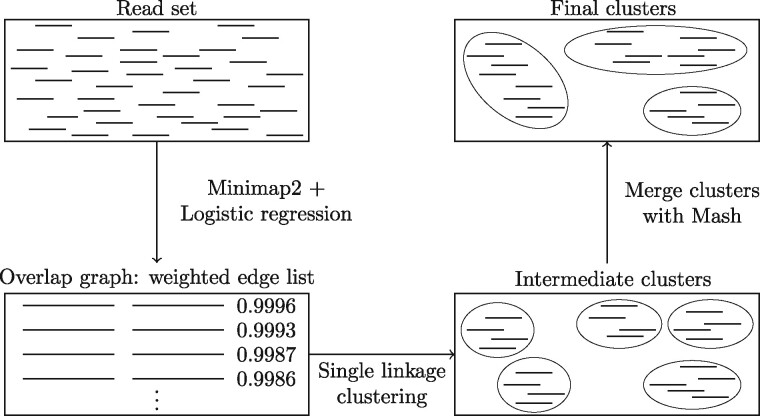
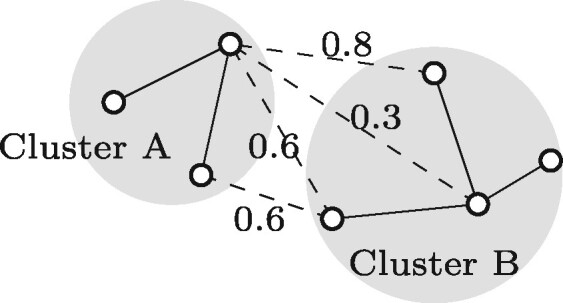
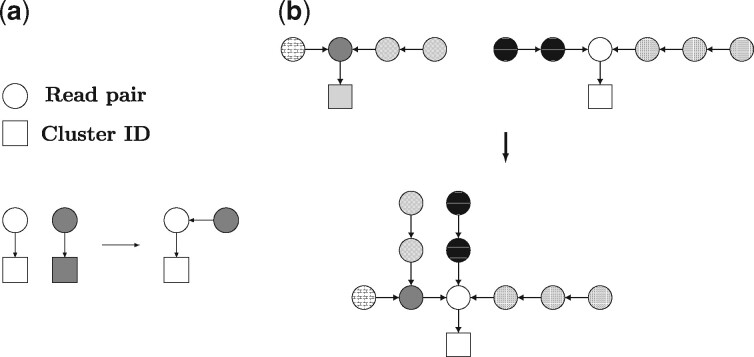
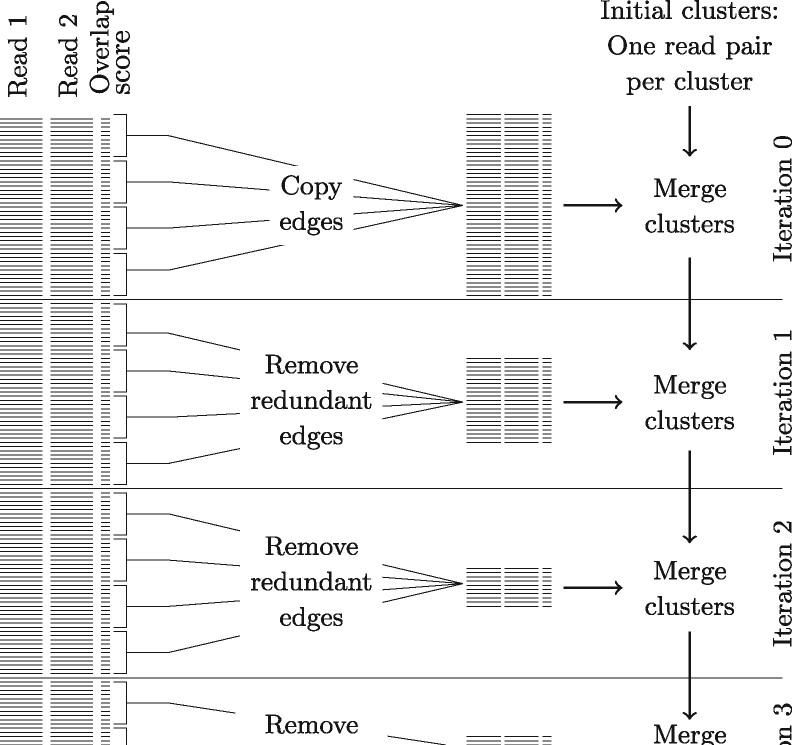
The microbes that live in an environment can be identified from the combined genomic material, also referred to as the metagenome. Sequencing a metagenome can result in large volumes of sequencing reads. A promising approach to reduce the size of metagenomic datasets is by clustering reads into groups based on their overlaps. Clustering reads are valuable to facilitate downstream analyses, including computationally intensive strain-aware assembly. As current read clustering approaches cannot handle the large datasets arising from high-throughput metagenome sequencing, a novel read clustering approach is needed. In this article, we propose OGRE, an Overlap Graph-based Read clustEring procedure for high-throughput sequencing data, with a focus on shotgun metagenomes.
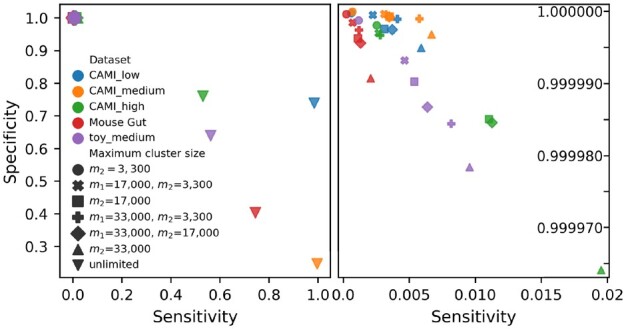
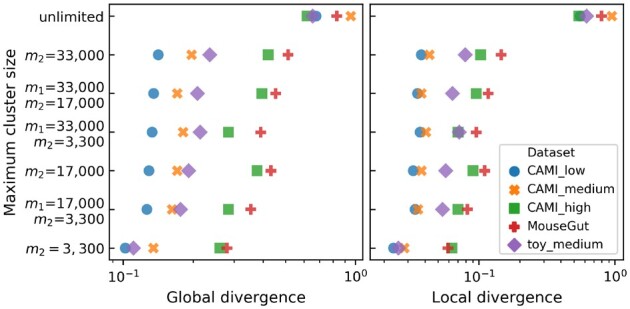
We show that for small datasets OGRE outperforms other read binners in terms of the number of species included in a cluster, also referred to as cluster purity, and the fraction of all reads that is placed in one of the clusters. Furthermore, OGRE is able to process metagenomic datasets that are too large for other read binners into clusters with high cluster purity.
OGRE is the only method that can successfully cluster reads in species-specific clusters for large metagenomic datasets without running into computation time- or memory issues.
Code is made available on Github (https://github.com/Marleen1/OGRE).
Supplementary data are available at Bioinformatics online.
生活在环境中的微生物可以通过组合基因组物质(也称为宏基因组)来识别。对宏基因组进行测序可能会产生大量的测序reads。一种很有前途的方法是通过基于重叠将reads 聚类成组来减小宏基因组数据集的大小。对 reads 进行聚类对于促进下游分析很有价值,包括计算密集型的基于菌株的组装。由于当前的 read 聚类方法无法处理来自高通量宏基因组测序的大型数据集,因此需要一种新的 read 聚类方法。在本文中,我们提出了 OGRE,这是一种基于重叠图的高通量测序数据 read 聚类程序,重点是shotgun 宏基因组。
我们表明,对于小数据集,OGRE 在聚类中包含的物种数量(也称为聚类纯度)和所有reads 中被放置在一个聚类中的部分方面优于其他 read 分类器。此外,OGRE 能够将其他 read 分类器无法处理的大型宏基因组数据集处理成具有高聚类纯度的聚类。
OGRE 是唯一一种能够在没有遇到计算时间或内存问题的情况下成功地将 reads 聚类到特定物种聚类中的方法,适用于大型宏基因组数据集。
代码可在 Github(https://github.com/Marleen1/OGRE)上获得。
补充数据可在 Bioinformatics 在线获得。