Molecular Genetics of Development Laboratory, Département des Sciences Biologiques, Université du Québec à Montréal (UQAM), Montréal, Québec, Canada.
Centre d'excellence en recherche sur les maladies orphelines-Fondation Courtois (CERMO-FC), Université du Québec à Montréal, Montréal, Québec, Canada.
PLoS Genet. 2020 Sep 8;16(9):e1009008. doi: 10.1371/journal.pgen.1009008. eCollection 2020 Sep.
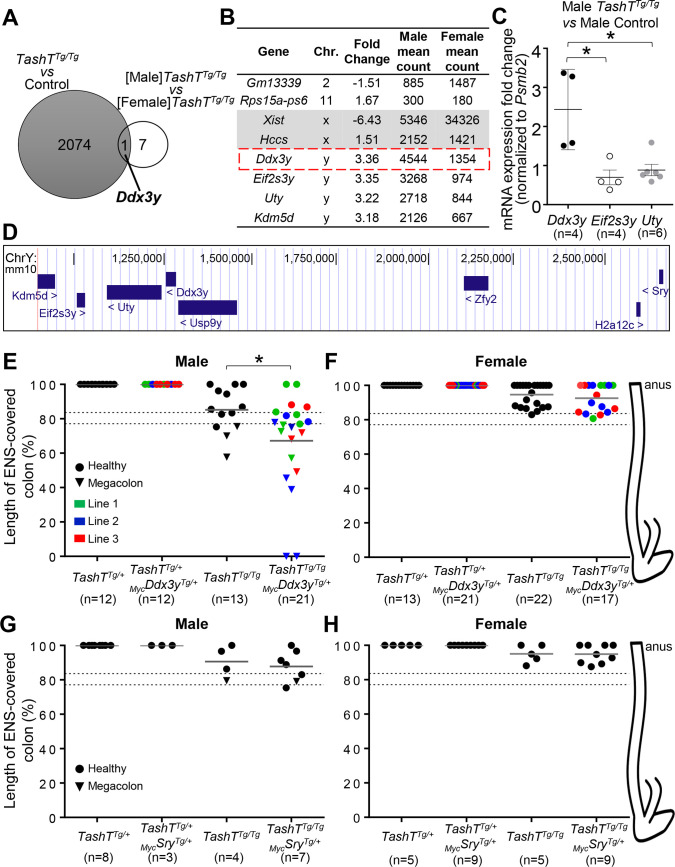
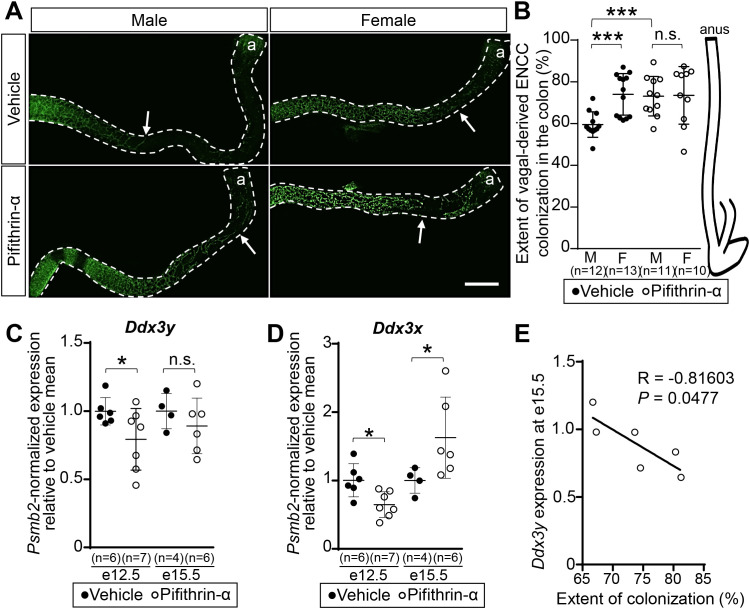
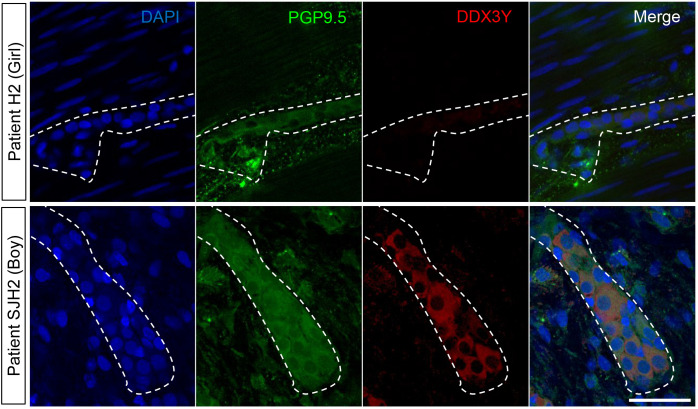
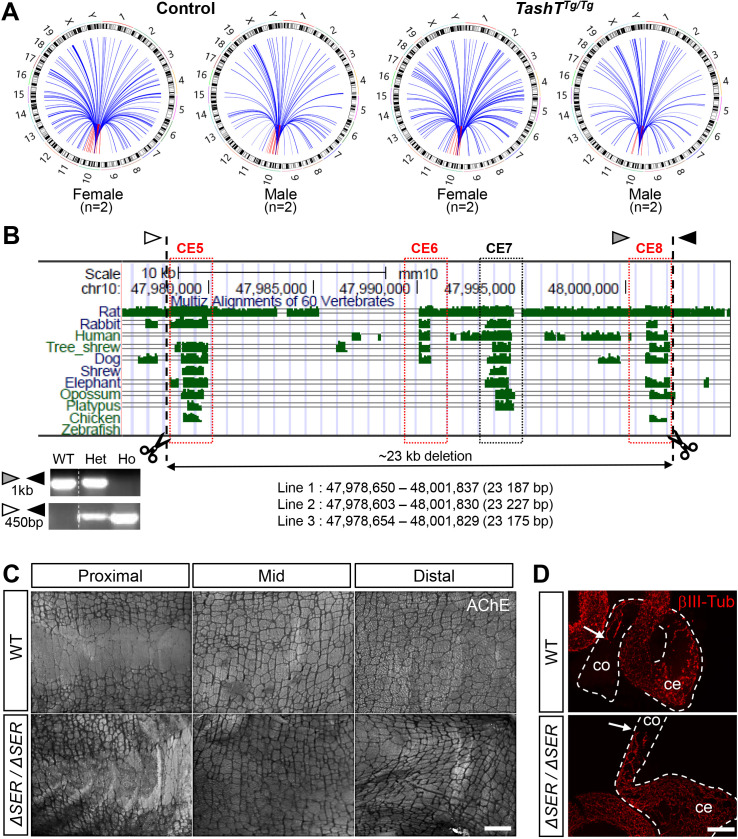
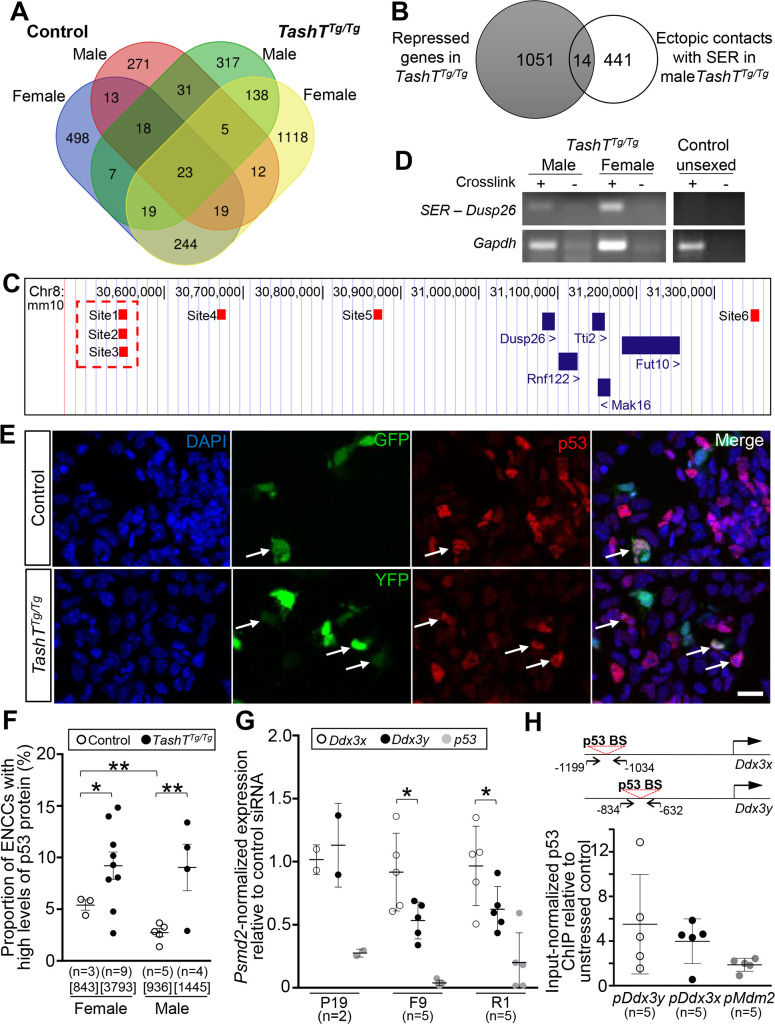
Hirschsprung disease (HSCR) is a complex genetic disorder of neural crest development resulting in incomplete formation of the enteric nervous system (ENS). This life-threatening neurocristopathy affects 1/5000 live births, with a currently unexplained male-biased ratio. To address this lack of knowledge, we took advantage of the TashT mutant mouse line, which is the only HSCR model to display a robust male bias. Our prior work revealed that the TashT insertional mutation perturbs a Chr.10 silencer-enriched non-coding region, leading to transcriptional dysregulation of hundreds of genes in neural crest-derived ENS progenitors of both sexes. Here, through sex-stratified transcriptome analyses and targeted overexpression in ENS progenitors, we show that male-biased ENS malformation in TashT embryos is not due to upregulation of Sry-the murine ortholog of a candidate gene for the HSCR male bias in humans-but instead involves upregulation of another Y-linked gene, Ddx3y. This discovery might be clinically relevant since we further found that the DDX3Y protein is also expressed in the ENS of a subset of male HSCR patients. Mechanistically, other data including chromosome conformation captured-based assays and CRISPR/Cas9-mediated deletions suggest that Ddx3y upregulation in male TashT ENS progenitors is due to increased transactivation by p53, which appears especially active in these cells yet without triggering apoptosis. Accordingly, in utero treatment of TashT embryos with the p53 inhibitor pifithrin-α decreased Ddx3y expression and abolished the otherwise more severe ENS defect in TashT males. Our data thus highlight novel pathogenic roles for p53 and DDX3Y during ENS formation in mice, a finding that might help to explain the intriguing male bias of HSCR in humans.
先天性巨结肠(HSCR)是一种复杂的神经嵴发育遗传疾病,导致肠神经系统(ENS)不完全形成。这种危及生命的神经嵴病变影响每 5000 例活产婴儿中就有 1 例,目前尚不清楚为什么存在男性偏倚的比例。为了解决这方面的知识空白,我们利用了 TashT 突变鼠系,这是唯一显示出强大男性偏倚的 HSCR 模型。我们之前的工作表明,TashT 插入突变会干扰富含 Chr.10 沉默子的非编码区域,导致雌雄两性神经嵴衍生的 ENS 祖细胞中数百个基因的转录失调。在这里,通过性别分层转录组分析和 ENS 祖细胞的靶向过表达,我们表明 TashT 胚胎中男性偏倚的 ENS 畸形不是由于 Sry 的上调——人类 HSCR 男性偏倚的候选基因的鼠类同源物——而是涉及另一个 Y 连锁基因 Ddx3y 的上调。这一发现可能具有临床相关性,因为我们进一步发现 DDX3Y 蛋白也在一组男性 HSCR 患者的 ENS 中表达。从机制上讲,其他数据包括染色体构象捕获分析和 CRISPR/Cas9 介导的缺失表明,雄性 TashT ENS 祖细胞中 Ddx3y 的上调是由于 p53 的转录激活增加所致,p53 在这些细胞中特别活跃,但不会引发细胞凋亡。因此,在 TashT 胚胎发育过程中,用 p53 抑制剂 pifithrin-α 进行宫内治疗,可降低 Ddx3y 的表达,并消除 TashT 雄性中更为严重的 ENS 缺陷。因此,我们的数据突出了 p53 和 DDX3Y 在小鼠 ENS 形成中的新的致病作用,这一发现可能有助于解释人类 HSCR 令人费解的男性偏倚。