Department of Neonatology, Jiaxing Maternity and Child Health Care Hospital, Jiaxing, China.
Department of Neonatology, Shanghai Children's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Biomed Res Int. 2020 Aug 31;2020:5690915. doi: 10.1155/2020/5690915. eCollection 2020.
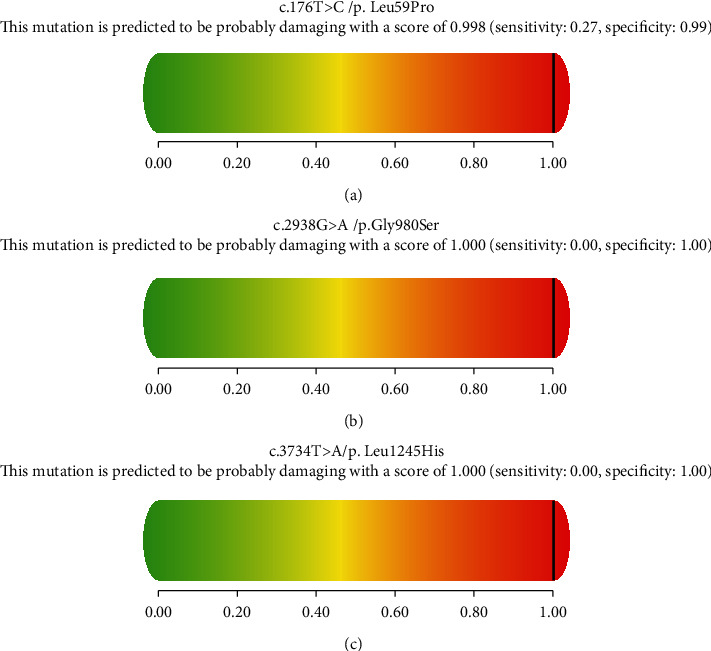
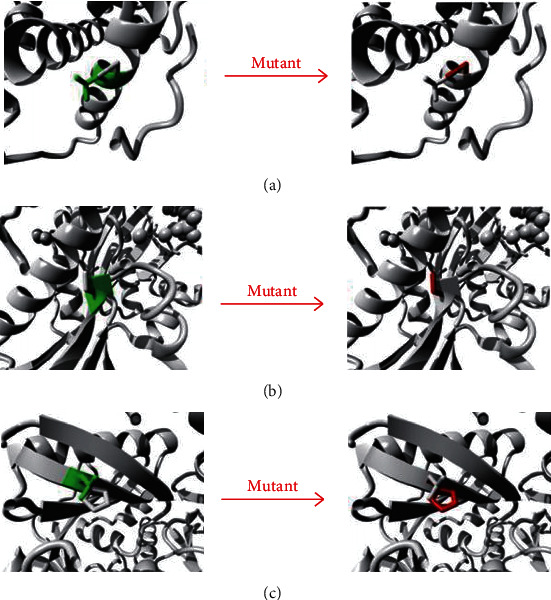
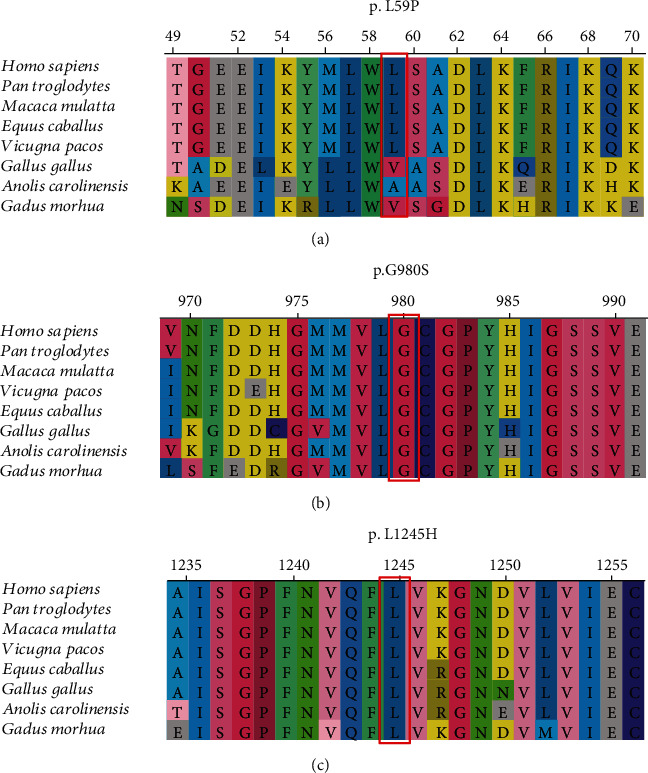
During Jan. 2016-Dec. 2019, nine Chinese patients from eight unrelated families were diagnosed with neonatal-onset UCDs by targeted panel sequencing or whole-exome sequencing (WES). Their clinical manifestations, biochemical features, 180-day-age outcomes, and molecular genetic characteristics were reviewed retrospectively. NGS-based tests revealed 7 patients diagnosed with ornithine transcarbamylase deficiency (OTCD) and 2 with carbamoylphosphate synthetase I deficiency (CPS1D). The spectrum of the clinical presentation of nine affected individuals progressed from unspecific symptoms like poor feeding to somnolence, coma, and death. All patients presented with an acute hyperammonemia. The most robust metabolic pattern in OTCD was hyperglutaminemic hyperammonemia with high concentration of urine orotic acid, and it was reported in six patients. Of ten variants found on the gene and CPS1 gene, 3 were novel: (c.176T>C (p.L59P)) in the gene, c.2938G>A (p.G980S) and c.3734T>A (p.L1245H) in the gene. There was a high mortality rate of 77.78% (7/9) for all the defects combined. An OTC-deficient male and a CPS1-deficient female survived from episodes of hyperammonemia. Although prompt recognition of UCD and the use of alternative pathway therapy in addition to provision of appropriate nutrition and dialysis improved survival, the overall outcomes for the neonatal-onset type are poor in China.
2016 年 1 月至 2019 年 12 月,通过靶向panel 测序或全外显子组测序(WES),诊断了 8 个无关家系的 9 例新生儿起病 UCD 患者。回顾性分析其临床表现、生化特征、180 天龄结局和分子遗传学特征。基于 NGS 的检测发现 7 例诊断为鸟氨酸转氨甲酰酶缺陷症(OTCD),2 例诊断为氨甲酰磷酸合成酶 I 缺陷症(CPS1D)。9 例受累个体的临床表现谱从非特异性症状(如喂养不良)进展为嗜睡、昏迷和死亡。所有患者均表现为急性高氨血症。OTCD 最显著的代谢特征是高谷氨酸血症伴高氨血症和高尿乳清酸浓度,6 例患者报告了该特征。在 基因和 CPS1 基因上发现的 10 个变异中,有 3 个是新的:基因上的 c.176T>C(p.L59P),基因上的 c.2938G>A(p.G980S)和 c.3734T>A(p.L1245H)。所有缺陷的总死亡率为 77.78%(7/9)。1 例 OTC 缺陷男性和 1 例 CPS1 缺陷女性从高氨血症发作中存活下来。尽管及时识别 UCD 并在提供适当营养和透析的基础上使用替代途径治疗,改善了生存,但中国新生儿起病型的总体预后仍较差。