Department of Genetics, University of Cambridge, Cambridge, CB3 0DH, UK.
Wellcome Sanger Institute, Cambridge, CB10 1SA, UK.
Genome Biol. 2020 Sep 17;21(1):250. doi: 10.1186/s13059-020-02160-7.
During the last decade, the analysis of ancient DNA (aDNA) sequence has become a powerful tool for the study of past human populations. However, the degraded nature of aDNA means that aDNA molecules are short and frequently mutated by post-mortem chemical modifications. These features decrease read mapping accuracy and increase reference bias, in which reads containing non-reference alleles are less likely to be mapped than those containing reference alleles. Alternative approaches have been developed to replace the linear reference with a variation graph which includes known alternative variants at each genetic locus. Here, we evaluate the use of variation graph software vg to avoid reference bias for aDNA and compare with existing methods.
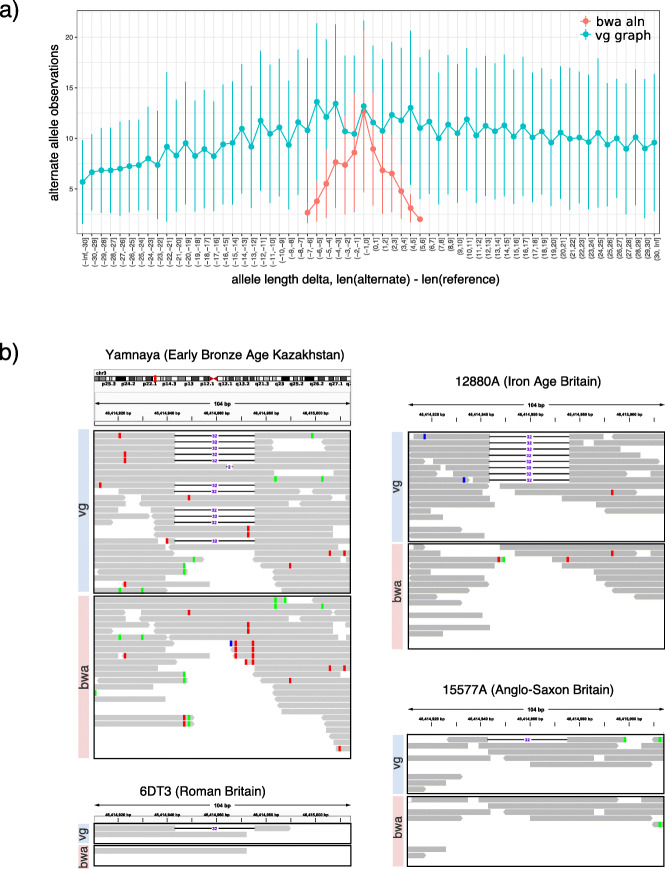
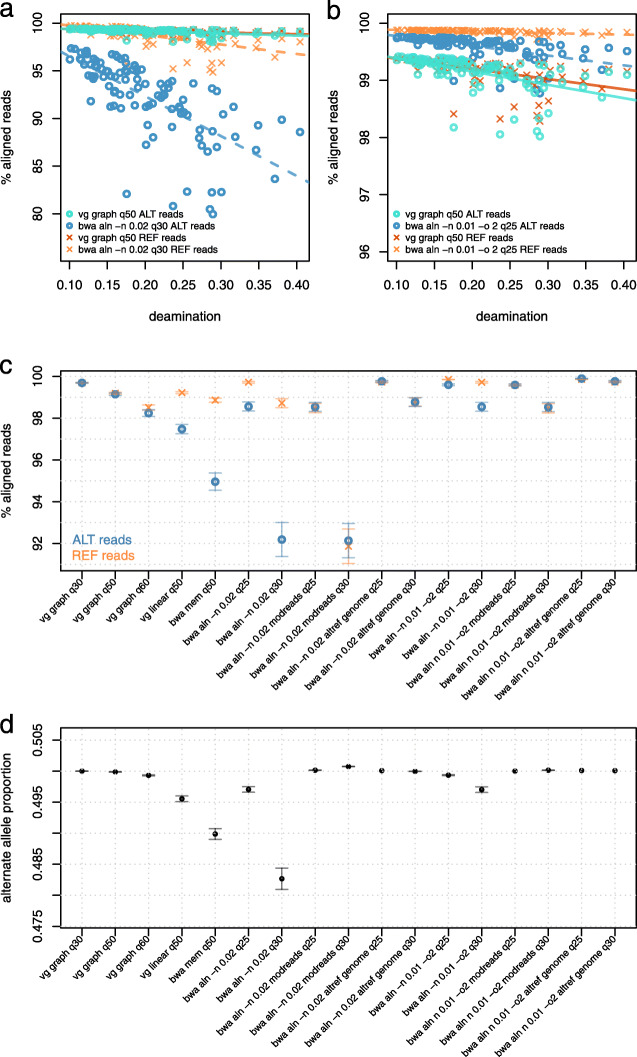
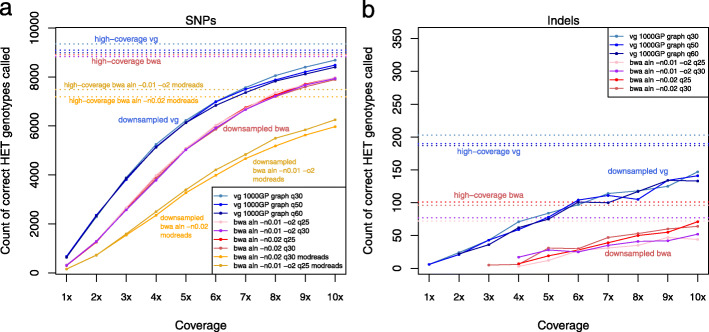
We use vg to align simulated and real aDNA samples to a variation graph containing 1000 Genome Project variants and compare with the same data aligned with bwa to the human linear reference genome. Using vg leads to a balanced allelic representation at polymorphic sites, effectively removing reference bias, and more sensitive variant detection in comparison with bwa, especially for insertions and deletions (indels). Alternative approaches that use relaxed bwa parameter settings or filter bwa alignments can also reduce bias but can have lower sensitivity than vg, particularly for indels.
Our findings demonstrate that aligning aDNA sequences to variation graphs effectively mitigates the impact of reference bias when analyzing aDNA, while retaining mapping sensitivity and allowing detection of variation, in particular indel variation, that was previously missed.
在过去的十年中,对古代 DNA(aDNA)序列的分析已成为研究过去人类群体的有力工具。然而,aDNA 的降解性质意味着 aDNA 分子较短,并且经常受到死后化学修饰的突变。这些特征降低了读取映射的准确性,并增加了参考偏差,其中包含非参考等位基因的读取比包含参考等位基因的读取更不可能被映射。已经开发了替代方法来用包含每个遗传基因座的已知替代变体的变异图代替线性参考。在这里,我们评估了使用变异图软件 vg 来避免 aDNA 的参考偏差,并与现有方法进行比较。
我们使用 vg 将模拟和真实的 aDNA 样本与包含 1000 个基因组计划变体的变异图对齐,并将其与用 bwa 对齐到人类线性参考基因组的相同数据进行比较。与 bwa 相比,使用 vg 可在多态性位点上实现平衡的等位基因表示,有效消除参考偏差,并提高变体检测的敏感性,尤其是对于插入和缺失(indels)。使用放宽 bwa 参数设置或过滤 bwa 比对的替代方法也可以减少偏差,但与 vg 相比,敏感性较低,特别是对于 indels。
我们的研究结果表明,将 aDNA 序列与变异图对齐可有效减轻分析 aDNA 时参考偏差的影响,同时保留映射敏感性,并允许检测以前错过的变异,特别是插入缺失(indel)变异。