Max Planck Institute for Evolutionary Anthropology, 04103, Leipzig, Germany.
BMC Biol. 2018 Oct 25;16(1):121. doi: 10.1186/s12915-018-0581-9.
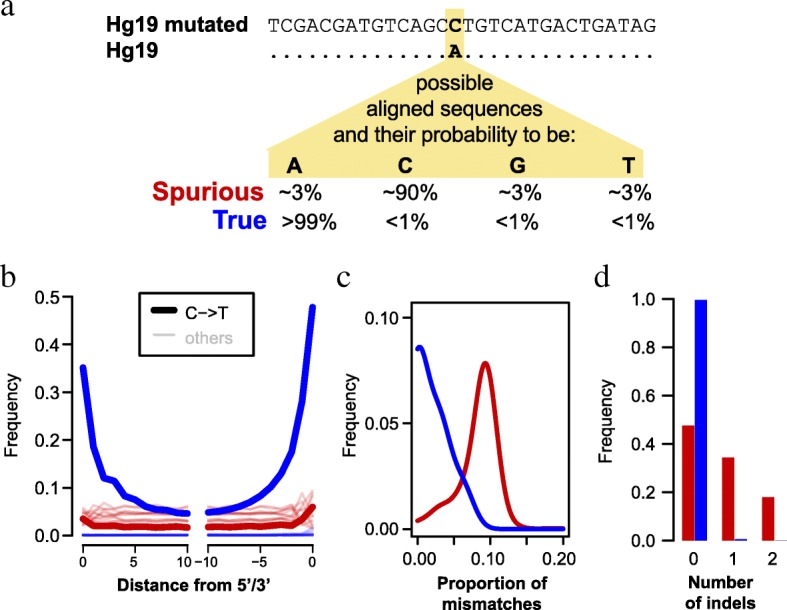
The study of ancient DNA is hampered by degradation, resulting in short DNA fragments. Advances in laboratory methods have made it possible to retrieve short DNA fragments, thereby improving access to DNA preserved in highly degraded, ancient material. However, such material contains large amounts of microbial contamination in addition to DNA fragments from the ancient organism. The resulting mixture of sequences constitutes a challenge for computational analysis, since microbial sequences are hard to distinguish from the ancient sequences of interest, especially when they are short.
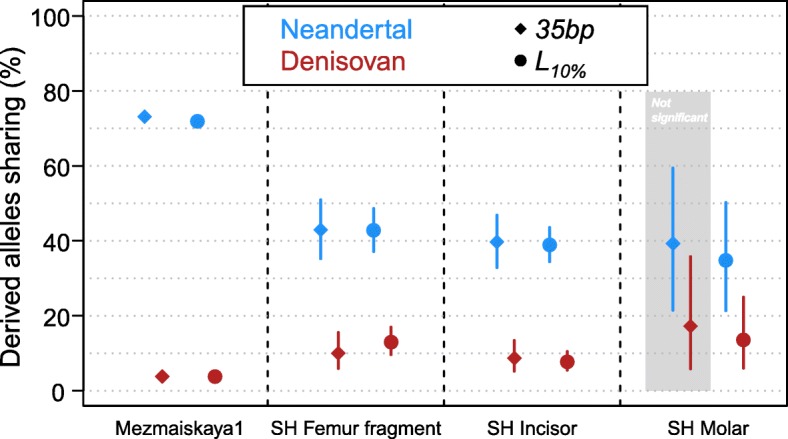
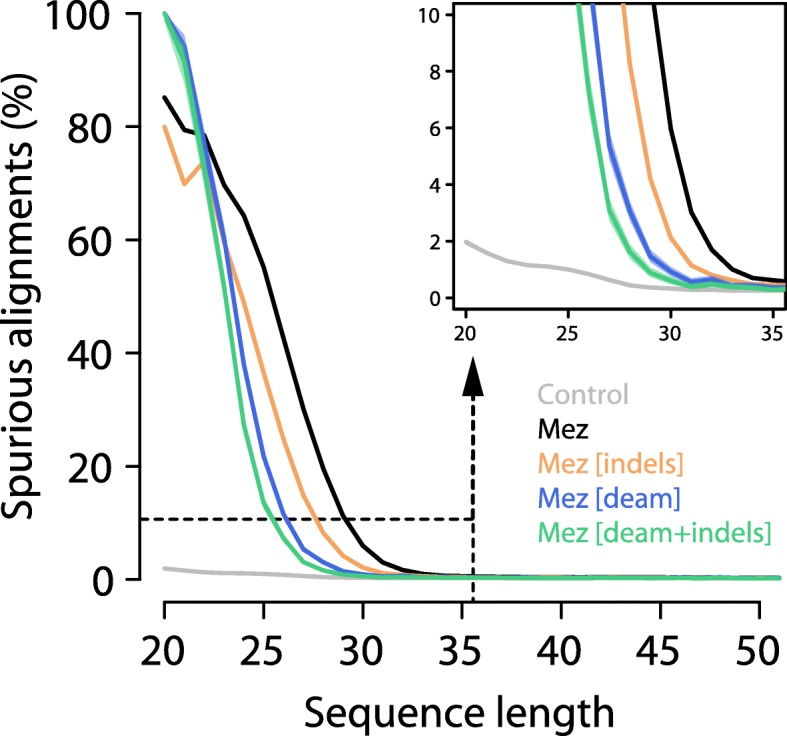
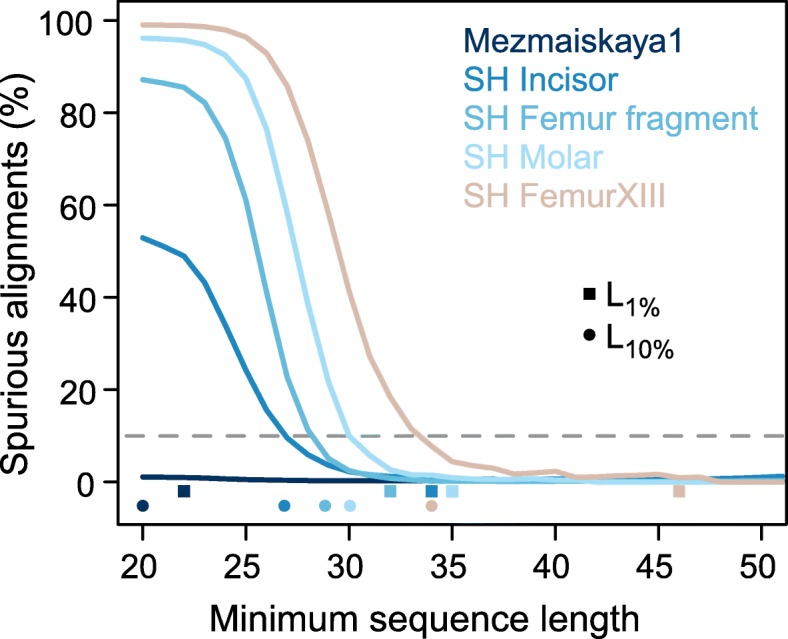
Here, we develop a method to quantify spurious alignments based on the presence or absence of rare variants. We find that spurious alignments are enriched for mismatches and insertion/deletion differences and lack substitution patterns typical of ancient DNA. The impact of spurious alignments can be reduced by filtering on these features and by imposing a sample-specific minimum length cutoff. We apply this approach to sequences from four ~ 430,000-year-old Sima de los Huesos hominin remains, which contain particularly short DNA fragments, and increase the amount of usable sequence data by 17-150%. This allows us to place a third specimen from the site on the Neandertal lineage.
Our method maximizes the sequence data amenable to genetic analysis from highly degraded ancient material and avoids pitfalls that are associated with the analysis of ultra-short DNA sequences.
古 DNA 的研究受到降解的阻碍,导致 DNA 片段较短。实验室方法的进步使得从高度降解的古代材料中提取短 DNA 片段成为可能,从而增加了对保存在其中的 DNA 的获取途径。然而,这种材料除了含有来自古老生物体的 DNA 片段外,还含有大量的微生物污染。由此产生的序列混合物对计算分析构成了挑战,因为微生物序列很难与感兴趣的古老序列区分开来,尤其是当它们很短时。
在这里,我们开发了一种基于稀有变异的存在或缺失来量化虚假比对的方法。我们发现,虚假比对富含错配和插入/缺失差异,并且缺乏古老 DNA 的典型替换模式。通过过滤这些特征并施加特定样本的最小长度截止值,可以减少虚假比对的影响。我们将这种方法应用于来自四个约 43 万年前的 Sima de los Huesos 古人类遗骸的序列中,这些遗骸中含有特别短的 DNA 片段,使可用序列数据量增加了 17% 到 150%。这使我们能够将该遗址的第三个标本置于尼安德特人谱系上。
我们的方法最大限度地提高了从高度降解的古代材料中进行遗传分析的序列数据量,并避免了与超短 DNA 序列分析相关的陷阱。