Pachajoa Harry, Claros-Hulbert Angelica, García-Quintero Ximena, Perafan Lina, Ramirez Andres, Zea-Vera Andres F
Faculty of Health Sciences, Congenital Anomalies and Rare Diseases Investigation Center (CIACER), Universidad Icesi, Cali, Colombia.
Genetic Department, Fundacion Valle del Lili, Cali, Colombia.
Appl Clin Genet. 2020 Sep 4;13:159-164. doi: 10.2147/TACG.S238715. eCollection 2020.
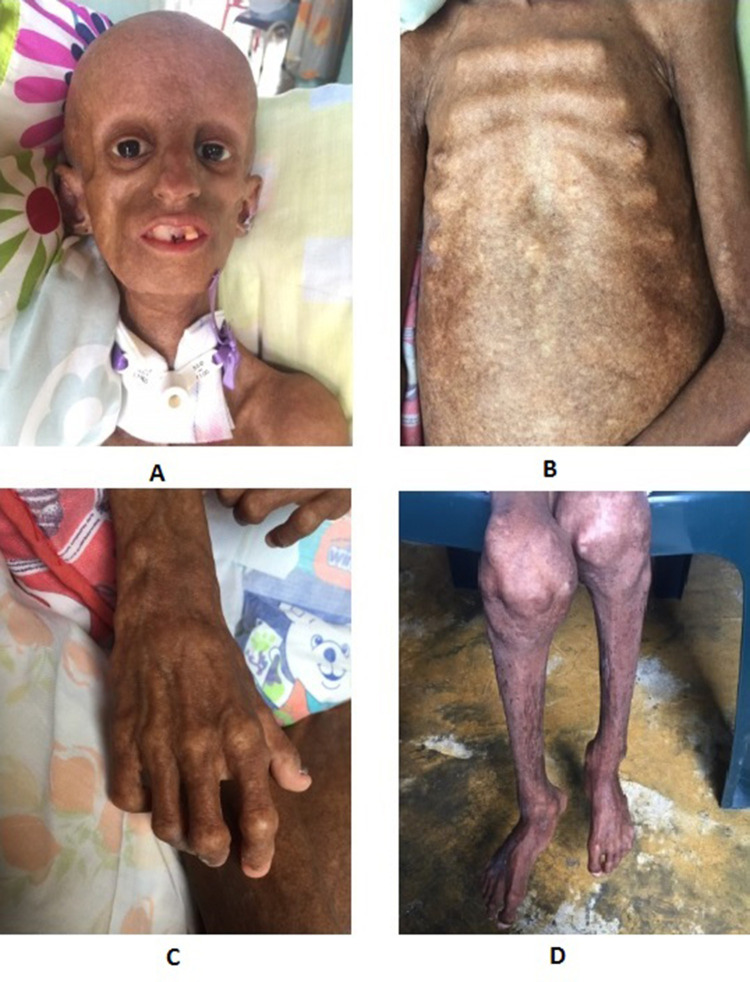
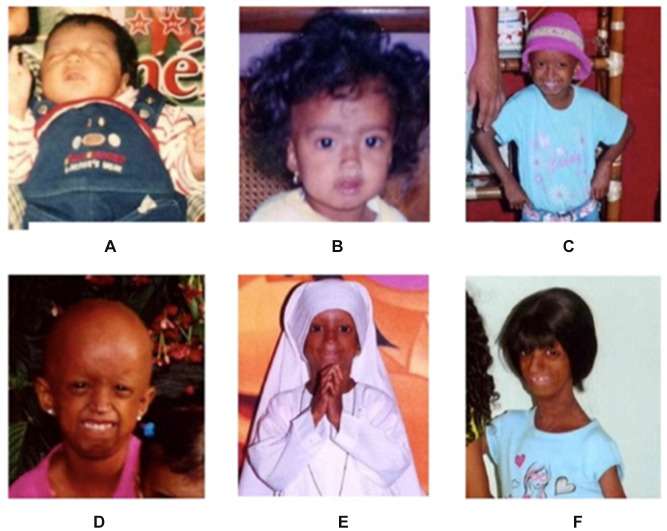
Hutchinson-Gilford progeria syndrome (HGPS) is a rare congenital disease caused by mutations in the gene. Children with HGPS are phenotypically characterized by lipodystrophy, short height, low body weight, scleroderma, reduced joint mobility, osteolysis, senile facial features, and cardiovascular compromise that usually lead to death. We aimed to describe the case of a patient who reached above-average age expectancy for children with HGPS in Latin America and describe the clinical and molecular characteristics of the patient. A 14-year-old female patient was presented with progeria-compatible phenotypic characteristics. HGPS was confirmed via gene sequencing that detected a heterozygous c.1824C>T (p.Gly608Gly) mutation. The primary aim is to describe the HGPS case, the molecular gene mutation finding, and make a short review of the limited available treatment options for children with HGPS. Such as the farnesyl transferase inhibitors in conjunction with other pharmacological therapies that have insinuated improvement in health, and survival rate.
哈钦森-吉尔福德早衰综合征(HGPS)是一种由该基因的突变引起的罕见先天性疾病。患有HGPS的儿童在表型上的特征为脂肪代谢障碍、身材矮小、体重低、硬皮病、关节活动度降低、骨质溶解、老年面部特征以及心血管功能损害,这些通常会导致死亡。我们旨在描述一名达到拉丁美洲患有HGPS儿童平均预期寿命以上的患者病例,并描述该患者的临床和分子特征。一名14岁女性患者表现出与早衰症相符的表型特征。通过基因测序确认了HGPS,该测序检测到一个杂合的c.1824C>T(p.Gly608Gly)突变。主要目的是描述HGPS病例、分子基因突变发现,并对患有HGPS儿童有限的可用治疗选择进行简要综述。例如法尼基转移酶抑制剂与其他暗示可改善健康和存活率的药物疗法联合使用。