Conradie Marrigje M
Department of Chemistry, University of the Free State, PO Box 339, 9300 Bloemfontein, Republic of South Africa.
Data Brief. 2020 Sep 1;32:106253. doi: 10.1016/j.dib.2020.106253. eCollection 2020 Oct.
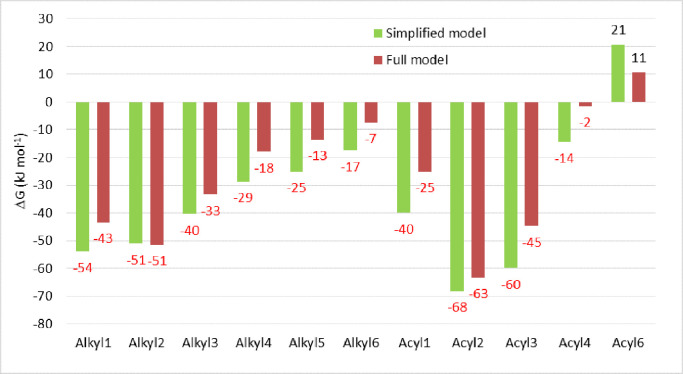
Density functional theory (DFT) free energy data and the reaction mechanism of the rhodium(triphenylphosphine)carbonyl-2,4-dioxo-3-pentyl-4-hydroxybenzoate plus iodomethane reaction are presented. The rhodium(I) reactant is a simplified model of the rhodium(I) of the rhodium(triphenylphosphine)carbonyl-2,4-dioxo-3-pentyl-4-decanyloxybenzoate plus iodomethane reaction (full model), presented in the related research article "Rhodium(triphenylphosphine)carbonyl-2,4-dioxo-3-pentyl-4-decanyloxybenzoate: A DFT study of Oxidative Addition and Methyl Migration" [1]. The goal is to illustrate that DFT calculations of a simplified model give the same information regarding the reaction scheme and free energy data as for the full model, while it requires much less computational resources to obtain the data. Furthermore the reaction scheme of the simplified model are in agreement with experimental observation of the full model [2].
本文给出了铑(三苯基膦)羰基 - 2,4 - 二氧代 - 3 - 戊基 - 4 - 羟基苯甲酸酯与碘甲烷反应的密度泛函理论(DFT)自由能数据及反应机理。铑(I)反应物是铑(三苯基膦)羰基 - 2,4 - 二氧代 - 3 - 戊基 - 4 - 癸氧基苯甲酸酯与碘甲烷反应(完整模型)中铑(I)的简化模型,该完整模型呈现在相关研究文章《铑(三苯基膦)羰基 - 2,4 - 二氧代 - 3 - 戊基 - 4 - 癸氧基苯甲酸酯:氧化加成和甲基迁移的DFT研究》[1]中。目的是说明简化模型的DFT计算能够给出与完整模型相同的关于反应方案和自由能数据的信息,同时获取这些数据所需的计算资源要少得多。此外,简化模型的反应方案与完整模型的实验观察结果一致[2]。