Department of Regenerative & Cancer Cell Biology, Albany Medical College, Albany, NY 12208, USA.
Cells. 2020 Sep 26;9(10):2172. doi: 10.3390/cells9102172.
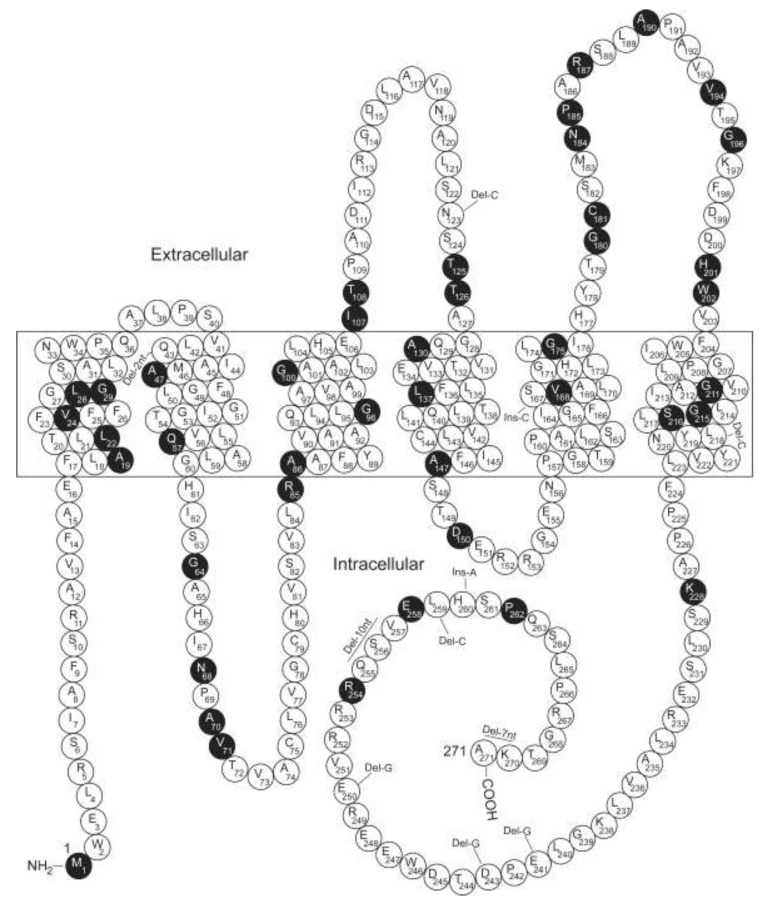
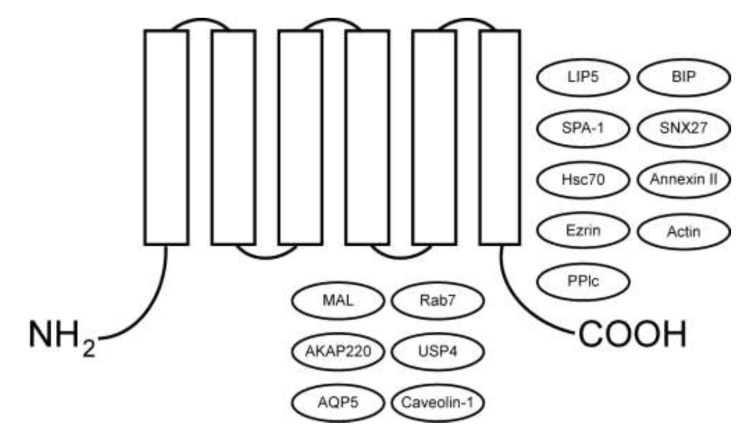
As a rare hereditary disease, congenital nephrogenic diabetes insipidus (NDI) is clinically characterized by polyuria with hyposthenuria and polydipsia. NDI results from collecting duct principal cell hyporesponsiveness or insensitivity to the antidiuretic action of arginine vasopressin (AVP). The principal cell-specific water channel aquaporin-2 (AQP2) plays an essential role in water reabsorption along osmotic gradients. The capacity to accumulate AQP2 in the apical plasma membrane in response to decreased fluid volume or increased plasma osmolality is critically regulated by the antidiuretic hormone AVP and its receptor 2 (AVPR2). Mutations in result in X-linked recessive NDI, the most common form of inherited NDI. Genetic defects in cause autosomal recessive or dominant NDI. In this review, we provide an updated overview of the genetic and molecular mechanisms of congenital NDI, with a focus on the potential disease-causing mutations in and , the molecular defects in the AVPR2 and AQP2 mutants, post-translational modifications (i.e., phosphorylation, ubiquitination, and glycosylation) and various protein-protein interactions that regulate phosphorylation, ubiquitination, tetramerization, trafficking, stability, and degradation of AQP2.
作为一种罕见的遗传性疾病,先天性肾性尿崩症(NDI)的临床特征为多尿伴尿比重降低和多饮。NDI 是由于集合管主细胞对血管加压素(AVP)的抗利尿作用反应迟钝或不敏感所致。主细胞特异性水通道 aquaporin-2(AQP2)在渗透压梯度下的水重吸收中起着至关重要的作用。AQP2 在细胞顶部质膜中的积累能力受抗利尿激素 AVP 和其受体 2(AVPR2)的严格调节。 中的突变导致 X 连锁隐性 NDI,这是最常见的遗传性 NDI 形式。 中的遗传缺陷导致常染色体隐性或显性 NDI。在这篇综述中,我们提供了先天性 NDI 的遗传和分子机制的最新概述,重点介绍了 和 中的潜在致病突变、AVPR2 和 AQP2 突变体的分子缺陷、翻译后修饰(即磷酸化、泛素化和糖基化)以及各种调节 AQP2 的磷酸化、泛素化、四聚体化、运输、稳定性和降解的蛋白-蛋白相互作用。