Department of Neurology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts, USA.
Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California, USA.
JCI Insight. 2020 Oct 2;5(19):136078. doi: 10.1172/jci.insight.136078.
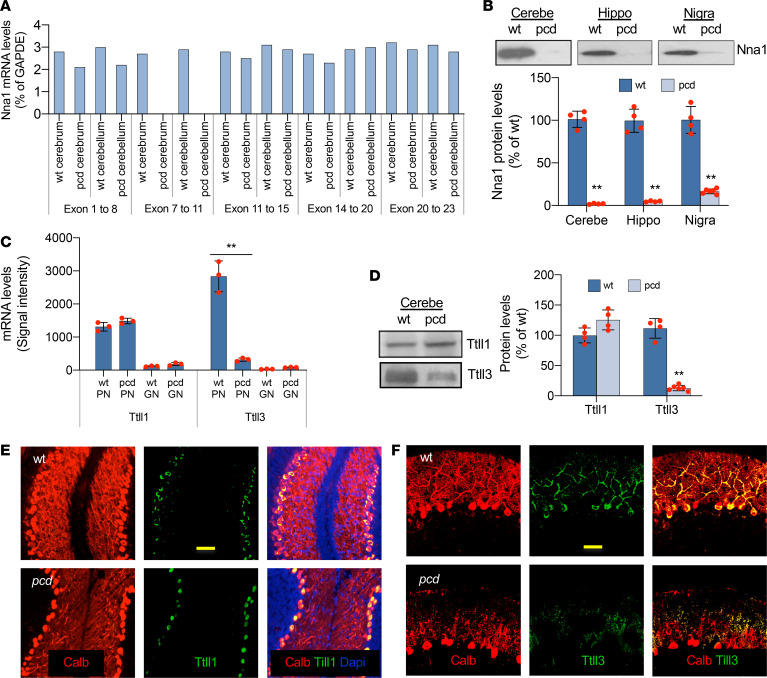
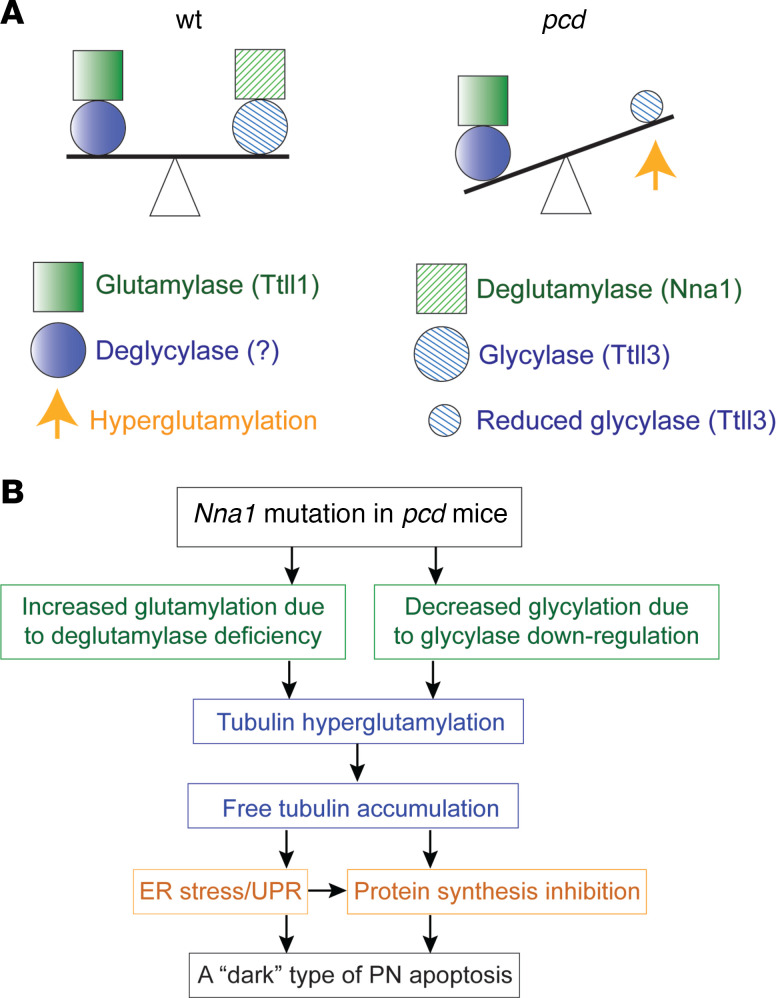
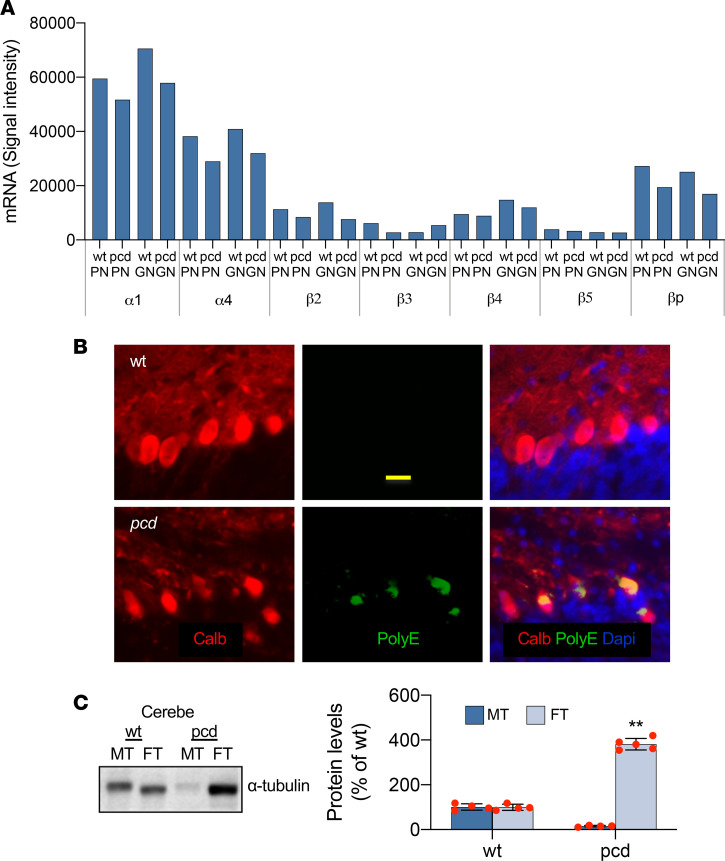
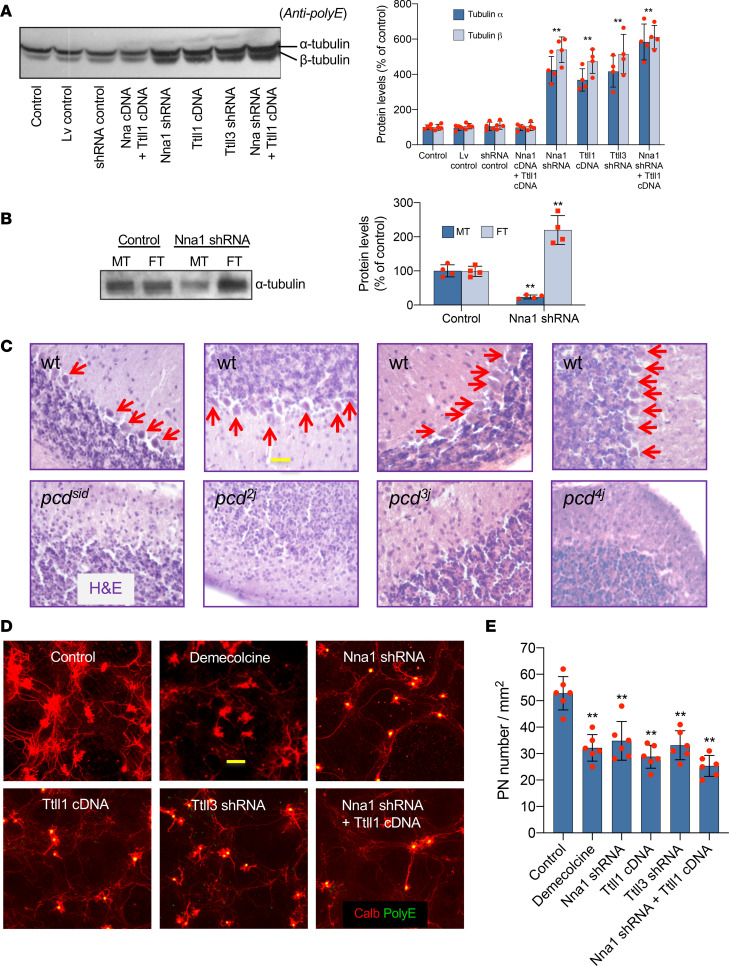
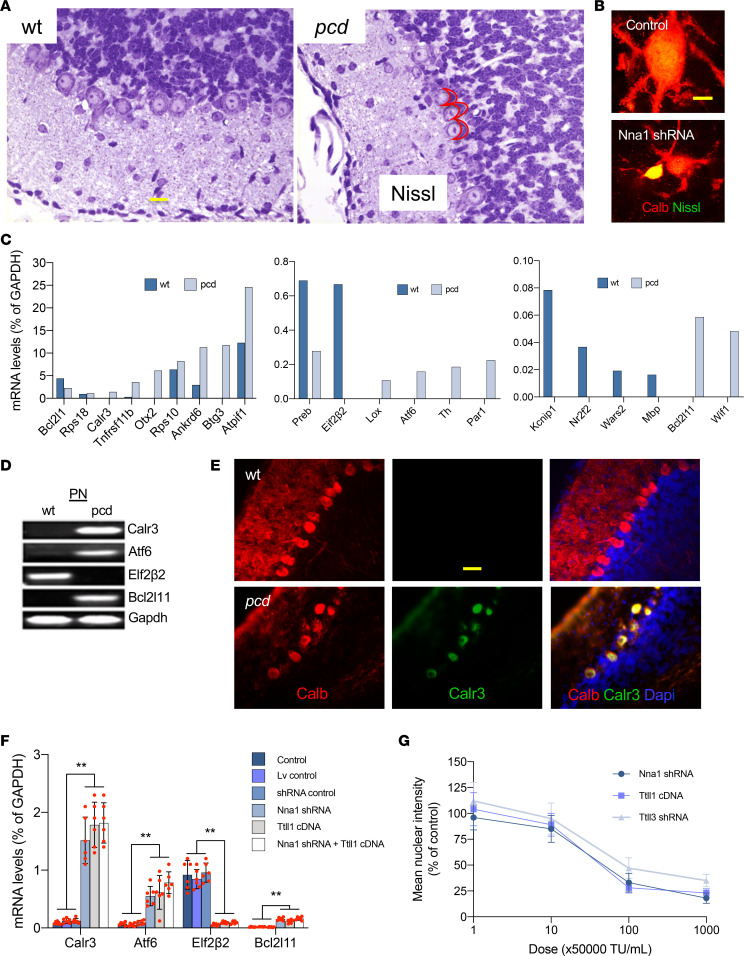
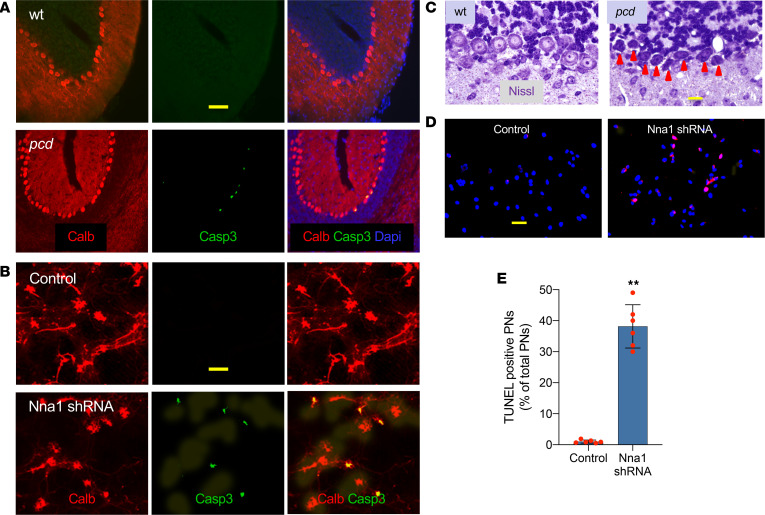
Posttranslational glutamylation/deglutamylation balance in tubulins influences dendritic maturation and neuronal survival of cerebellar Purkinje neurons (PNs). PNs and some additional neuronal types degenerate in several spontaneous, independently occurring Purkinje cell degeneration (pcd) mice featuring mutant neuronal nuclear protein induced by axotomy (Nna1), a deglutamylase gene. This defective deglutamylase allows glutamylases to form hyperglutamylated tubulins. In pcd, all PNs die during postnatal "adolescence." Neurons in some additional brain regions also die, mostly later than PNs. We show in laser capture microdissected single PNs, in cerebellar granule cell neuronal clusters, and in dissected hippocampus and substantia nigra that deglutamase mRNA and protein were virtually absent before pcd PNs degenerated, whereas glutaminase mRNA and protein remained normal. Hyperglutamylated microtubules and dimeric tubulins accumulated in pcd PNs and were involved in pcd PN death by glutamylase/deglutamylase imbalance. Importantly, treatment with a microtubule depolymerizer corrected the glutamylation/deglutamylation ratio, increasing PN survival. Further, before onset of neuronal death, pcd PNs displayed prominent basal polylisosomal masses rich in ER. We propose a "seesaw" metamorphic model summarizing mutant Nna1-induced tubulin hyperglutamylation, the pcd's PN phenotype, and report that the neuronal disorder involved ER stress, unfolded protein response, and protein synthesis inhibition preceding PN death by apoptosis/necroptosis.
微管蛋白的翻译后谷氨酰化/去谷氨酰化平衡影响小脑浦肯野神经元(PNs)的树突成熟和神经元存活。在几种自发出现的、独立发生的浦肯野细胞变性(pcd)小鼠中,PNs和其他一些神经元类型会发生退化,这些小鼠具有由轴突切断诱导的突变神经元核蛋白(Nna1),一种去谷氨酰酶基因。这种有缺陷的去谷氨酰酶使谷氨酰酶形成高度谷氨酰化的微管蛋白。在pcd小鼠中,所有PNs在出生后的“青春期”死亡。其他一些脑区的神经元也会死亡,大多比PNs死亡时间晚。我们在激光捕获显微切割的单个PNs、小脑颗粒细胞神经元簇以及解剖的海马体和黑质中发现,在pcd PNs退化之前,去谷氨酰酶mRNA和蛋白几乎不存在,而谷氨酰胺酶mRNA和蛋白保持正常。高度谷氨酰化的微管和二聚体微管蛋白在pcd PNs中积累,并通过谷氨酰酶/去谷氨酰酶失衡参与pcd PN死亡。重要的是,用微管解聚剂治疗可纠正谷氨酰化/去谷氨酰化比率,提高PNs的存活率。此外,在神经元死亡开始之前,pcd PNs显示出富含内质网的突出的基底多聚核糖体团块。我们提出了一个“跷跷板”变质模型,总结了突变Nna1诱导的微管蛋白高度谷氨酰化、pcd的PN表型,并报告神经元疾病涉及内质网应激、未折叠蛋白反应以及在PNs通过凋亡/坏死性凋亡死亡之前的蛋白质合成抑制。