Division of Hematology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
Van Creveldkliniek, University Medical Centre Utrecht, University of Utrecht, Utrecht, the Netherlands.
Haematologica. 2020 Sep 1;105(9):2229-2239. doi: 10.3324/haematol.2019.240846.
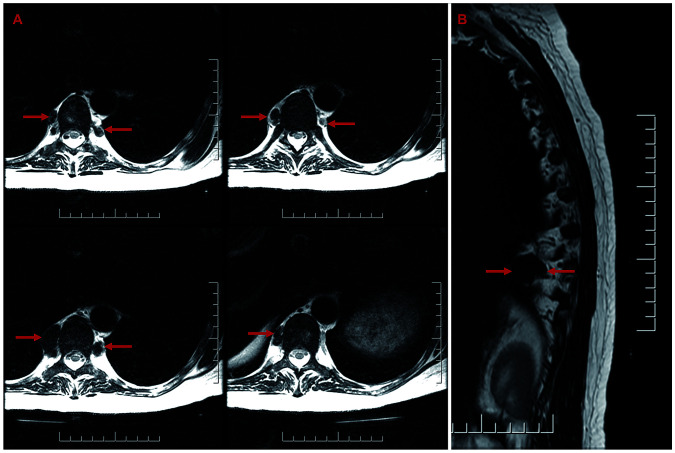
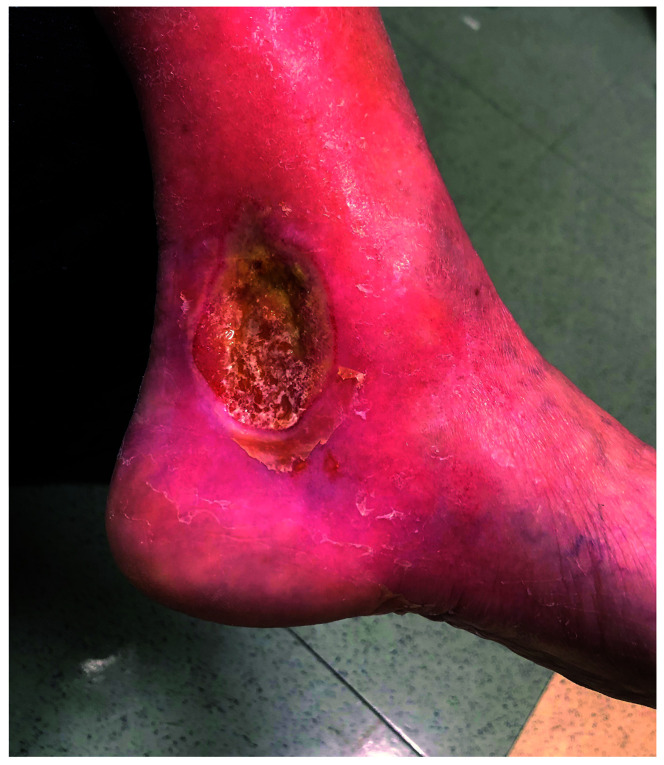
Pyruvate kinase deficiency (PKD) is the most common cause of chronic hereditary non-spherocytic hemolytic anemia and results in a broad spectrum of disease. The diagnosis of PKD requires a high index of suspicion and judicious use of laboratory tests that may not always be informative, including pyruvate kinase enzyme assay and genetic analysis of the PKLR gene. A significant minority of patients with PKD have occult mutations in non-coding regions of PKLR which are missed on standard genetic tests. The biochemical consequences of PKD result in hemolytic anemia due to red cell pyruvate and ATP deficiency while simultaneously causing increased red cell 2,3-diphosphoglycerate, which facilitates oxygen unloading. This phenomenon, in addition to numerous other factors such as genetic background and differences in splenic function result in a poor correlation between symptoms and degree of anemia from patient to patient. Red cell transfusions should, therefore, be symptom-directed and not based on a hemoglobin threshold. Patients may experience specific complications, such as paravertebral extramedullary hematopoiesis and chronic debilitating icterus, which require personalized treatment. The decision to perform splenectomy or hematopoietic stem cell transplantation is nuanced and depends on disease burden and long-term outlook given that targeted therapeutics are in development. In recognition of the complicated nature of the disease and its management and the limitations of the PKD literature, an international working group of ten PKD experts convened to better define the disease burden and manifestations. This article summarizes the conclusions of this working group and is a guide for clinicians and investigators caring for patients with PKD.
丙酮酸激酶缺乏症(PKD)是慢性遗传性非球形红细胞溶血性贫血的最常见原因,导致广泛的疾病谱。PKD 的诊断需要高度怀疑和明智地使用实验室检查,这些检查并不总是具有信息性,包括丙酮酸激酶酶测定和 PKLR 基因的遗传分析。少数 PKD 患者存在非编码区隐匿性突变,标准遗传检测会遗漏这些突变。PKD 的生化后果导致红细胞丙酮酸和 ATP 缺乏引起溶血性贫血,同时导致红细胞 2,3-二磷酸甘油酸增加,从而促进氧释放。除了遗传背景和脾功能差异等许多其他因素外,这种现象导致患者之间的症状和贫血程度之间相关性较差。因此,红细胞输血应针对症状,而不是基于血红蛋白阈值。患者可能会出现特定的并发症,如椎旁髓外造血和慢性虚弱性黄疸,需要个体化治疗。是否进行脾切除术或造血干细胞移植的决定是微妙的,取决于疾病负担和长期预后,因为正在开发靶向治疗方法。鉴于该疾病及其管理的复杂性以及 PKD 文献的局限性,一个由 10 名 PKD 专家组成的国际工作组召开会议,以更好地定义疾病负担和表现。本文总结了该工作组的结论,是照顾 PKD 患者的临床医生和研究人员的指南。