Mendonça Rodrigo de Holanda, Matsui Ciro, Polido Graziela Jorge, Silva André Macedo Serafim, Kulikowski Leslie, Torchio Dias Alexandre, Zanardo Evelin Aline, Solla Davi Jorge Fontoura, Gurgel-Giannetti Juliana, de Moura Ana Carolina Monteiro Lessa, Sampaio Gabriela Palhares Campolina, Oliveira Acary Souza Bulle, de Souza Paulo Victor Sgobbi, Pinto Wladimir Bocca Vieira de Rezende, Gonçalves Eduardo Augusto, Farias Igor Braga, Nardes Flávia, Araújo Alexandra Prufer de Queiroz Campos, Marques Wilson, Tomaselli Pedro José, Ribeiro Mara Dell Ospedale, Kitajima João Paulo, Paoli Monteiro Fabíola, Saute Jonas Alex Morales, Becker Michele Michelin, Saraiva-Pereira Maria Luiza, Brusius-Facchin Ana Carolina, van der Linden Vanessa, Florêncio Rodrigo Neves, Barbosa André Vinícius Soares, Machado-Costa Marcela Camara, Pessoa André Luiz Santos, Souza Leticia Silva, Franca Marcondes Cavalcante, Kok Fernando, Reed Umbertina Conti, Zanoteli Edmar
Department of Neurology (R.H.M., C.M., G.J.P., A.M.S.S., D.J.F.S., F.K., U.C.R., E.Z.); Department of Pathology (L.K., A.T.D., E.A.Z.), Faculdade de Medicina da Universidade de São Paulo (FMUSP); Departamento de Pediatria e Neuropediatria (J.G.-G., A.C.M.L.M., G.P.C.S.), Hospital das Clínicas da Universidade Federal de Minas Gerais, Belo Horizonte; Departamento de Neurologia - UNIFESP (A.S.B.O., P.V.S.S., W.B.V.R.P., E.A.G., I.B.F.), São Paulo; Departamento de Pediatria, Seção de Neurologia Infantil - UFRJ (F.N., A.P.Q.C.A.), Rio de Janeiro; Departamento de Neurologia (W.M., P.J.T.), FMUSP-RP, Ribeirao Preto; Mendelics Análise Genômica (M.D.O.R., J.P.K., F.P.M., F.K.), São Paulo; Serviço de Neurologia (J.A.M.S.), Hospital de Clinicas de Porto Alegre, Universidade Federal do Rio Grande do Sul, UFRGS, Porto Alegre; Unidade de Neurologia Infantil (M.M.B.), Hospital de Clinicas de Porto Alegre; Serviço de Genética Médica (J.A.M.S., M.L.S.-P., A.C.B.-F.), Hospital de Clinicas de Porto Alegre; UFRGS, Porto Alegre; Departamento de Bioquímica - UFRGS (M.L.S.-P.), Porto Alegre; Hospital Maria Lucinda (V.L., R.N.F.), Recife; Hospital Infantil Joao Paulo II (A.V.S.B.), Fundação Hospitalar de Minas Gerais, Belo Horizonte; Escola Bahiana de Medicina e Saúde Pública (M.C.M.-C.), Salvador; Hospital Infantil Albert Sabin (A.L.S.P.), Universidade Estadual do Ceará, Fortaleza; and Departamento de Neurologia (L.S.S., M.C.F.), Unicamp, Campinas, Brazil.
Neurol Genet. 2020 Sep 1;6(5):e505. doi: 10.1212/NXG.0000000000000505. eCollection 2020 Oct.
The aim of the study was to report the proportion of homozygous and compound heterozygous variants in the survival motor neuron 1 () gene in a large population of patients with spinal muscular atrophy (SMA) and to correlate the severity of the disease with the presence of specific intragenic variants in and with the copy number.
Four hundred fifty Brazilian patients with SMA were included in a retrospective study, and clinical data were analyzed compared with genetic data; the copy number was obtained by multiplex ligation-dependent probe amplification and pathogenic variants in by next-generation sequencing.
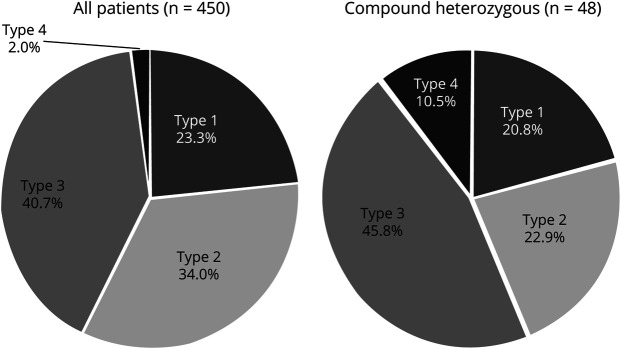
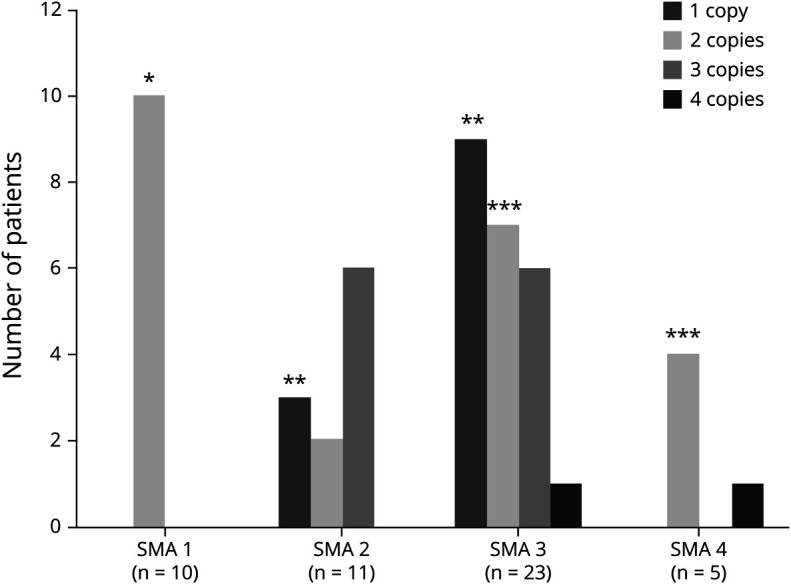
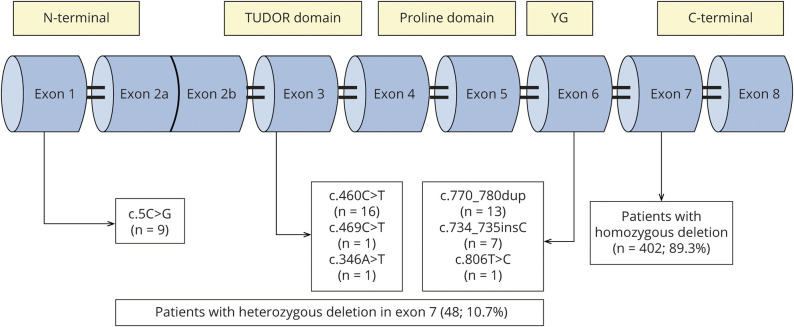
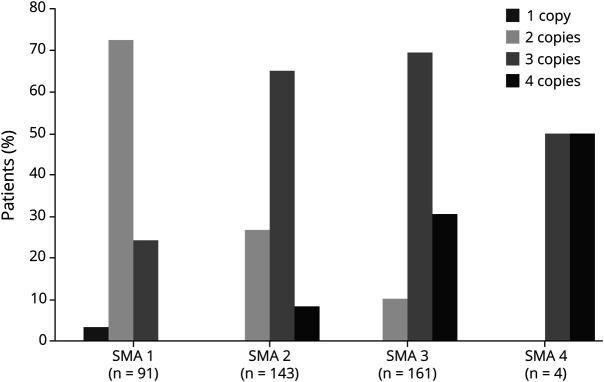
Four hundred two patients (89.3%) presented homozygous exon 7- deletion, and 48 (10.7%) were compound heterozygous for the common deletion in one allele and a point mutation in the other allele. Recurrent variants in exons 3 and 6 (c.460C>T, c.770_780dup and c.734_735insC) accounted for almost 80% of compound heterozygous patients. Another recurrent pathogenic variant was c.5C>G at exon 1. Patients with c.770_780dup and c.734_735insC had a clinical phenotype correlated with copy number, whereas the variants c.460C>T and c.5C>G determined a milder phenotype independently of the copies.
Patients with specific pathogenic variants (c.460C>T and c.5C>G) presented a milder phenotype, and the copy number did not correlate with disease severity in this group.
本研究旨在报告大量脊髓性肌萎缩症(SMA)患者中生存运动神经元1(SMN1)基因纯合和复合杂合变异的比例,并将疾病严重程度与SMN1基因中特定基因内变异的存在以及SMN2拷贝数相关联。
450例巴西SMA患者纳入一项回顾性研究,分析临床数据并与基因数据进行比较;通过多重连接依赖探针扩增获得SMN2拷贝数,通过下一代测序获得SMN1基因中的致病变异。
402例患者(89.3%)呈现纯合的外显子7缺失,48例(10.7%)为一个等位基因常见缺失且另一个等位基因存在点突变的复合杂合子。外显子3和6中的复发性变异(c.460C>T、c.770_780dup和c.734_735insC)占复合杂合患者的近80%。另一个复发性致病变异是外显子1的c.5C>G。携带c.770_780dup和c.734_735insC的患者临床表型与SMN2拷贝数相关,而c.460C>T和c.5C>G变异独立于SMN2拷贝数决定较轻的表型。
具有特定致病变异(c.460C>T和c.5C>G)的患者呈现较轻的表型,且该组患者的SMN2拷贝数与疾病严重程度无关。