Taylor Alison M, Macari Elizabeth R, Chan Iris T, Blair Megan C, Doulatov Sergei, Vo Linda T, Raiser David M, Siva Kavitha, Basak Anindita, Pirouz Mehdi, Shah Arish N, McGrath Katherine, Humphries Jessica M, Stillman Emma, Alter Blanche P, Calo Eliezer, Gregory Richard I, Sankaran Vijay G, Flygare Johan, Ebert Benjamin L, Zhou Yi, Daley George Q, Zon Leonard I
Stem Cell Program, Boston Children's Hospital and Harvard Stem Cell Institute, Boston, MA 02115, USA.
Division of Hematology/Oncology, Boston Children's Hospital and Dana Farber Cancer Institute, Boston, MA 02115, USA.
Sci Transl Med. 2020 Oct 21;12(566). doi: 10.1126/scitranslmed.abb5831.
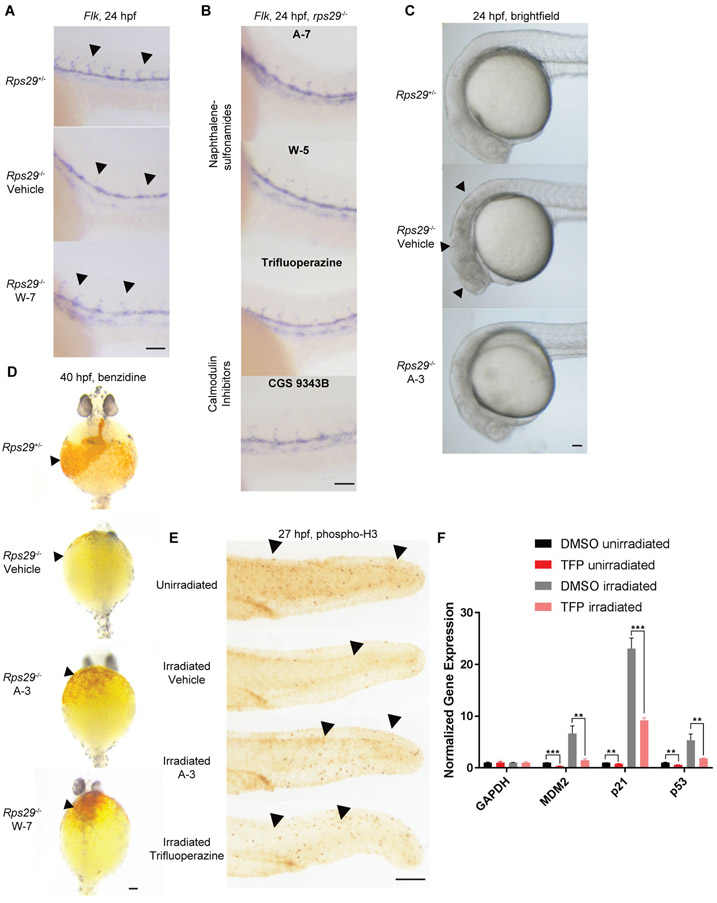
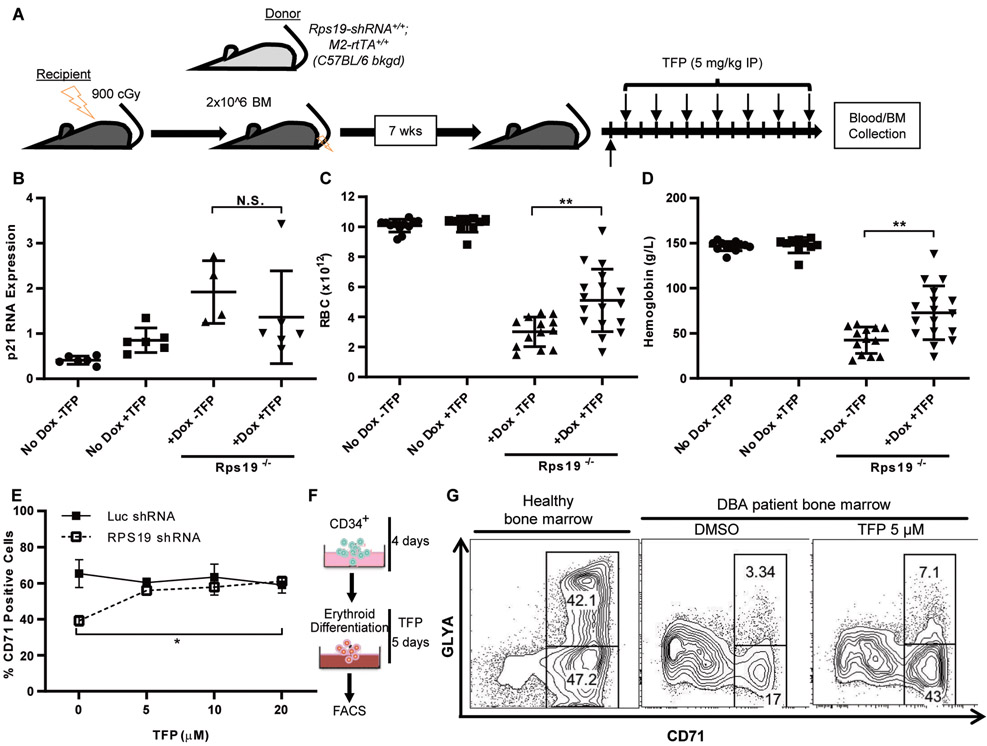
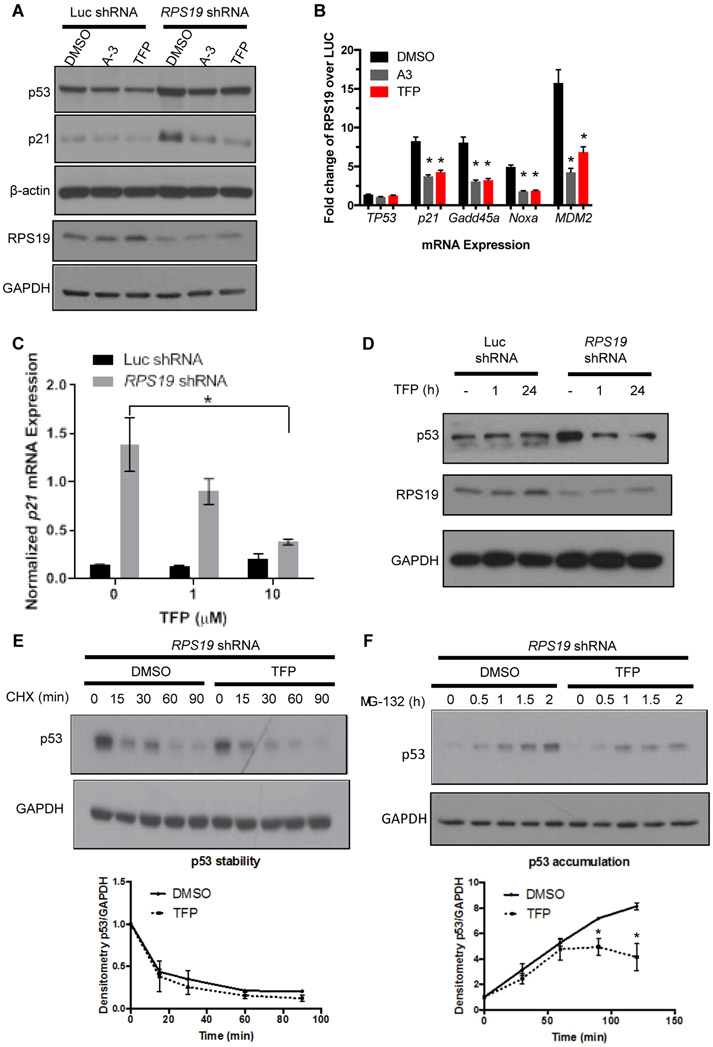
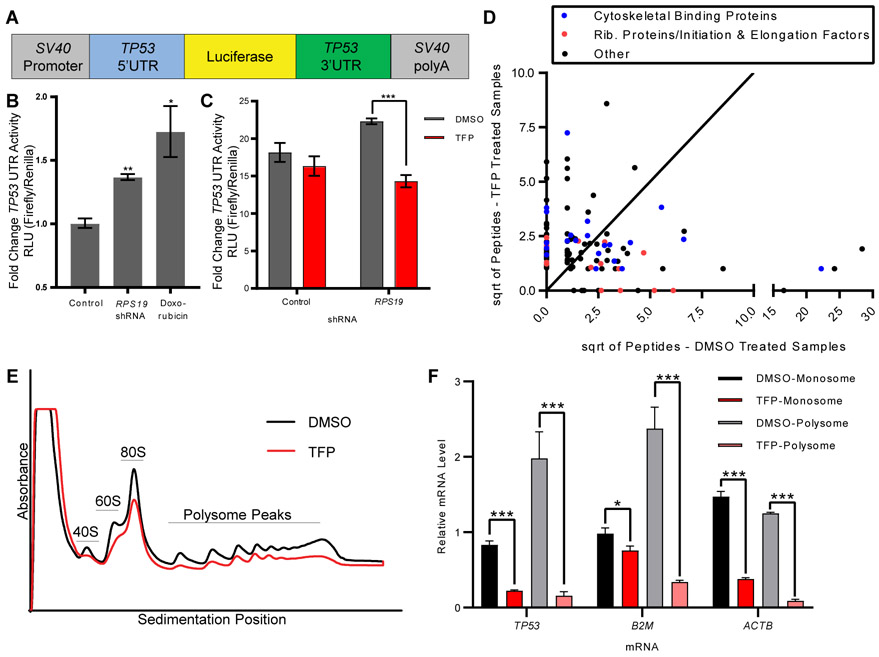
Diamond-Blackfan anemia (DBA) is a rare hematopoietic disease characterized by a block in red cell differentiation. Most DBA cases are caused by mutations in ribosomal proteins and characterized by higher than normal activity of the tumor suppressor p53. Higher p53 activity is thought to contribute to DBA phenotypes by inducing apoptosis during red blood cell differentiation. Currently, there are few therapies available for patients with DBA. We performed a chemical screen using zebrafish ribosomal small subunit protein 29 () mutant embryos that have a p53-dependent anemia and identified calmodulin inhibitors that rescued the phenotype. Our studies demonstrated that calmodulin inhibitors attenuated p53 protein amount and activity. Treatment with calmodulin inhibitors led to decreased p53 translation and accumulation but does not affect p53 stability. A U.S. Food and Drug Administration-approved calmodulin inhibitor, trifluoperazine, rescued hematopoietic phenotypes of DBA models in vivo in zebrafish and mouse models. In addition, trifluoperazine rescued these phenotypes in human CD34 hematopoietic stem and progenitor cells. Erythroid differentiation was also improved in CD34 cells isolated from a patient with DBA. This work uncovers a potential avenue of therapeutic development for patients with DBA.
先天性纯红细胞再生障碍性贫血(DBA)是一种罕见的造血疾病,其特征是红细胞分化受阻。大多数DBA病例由核糖体蛋白突变引起,其特征是肿瘤抑制因子p53的活性高于正常水平。较高的p53活性被认为通过在红细胞分化过程中诱导细胞凋亡而导致DBA表型。目前,针对DBA患者的治疗方法很少。我们使用具有p53依赖性贫血的斑马鱼核糖体小亚基蛋白29()突变胚胎进行了化学筛选,并鉴定出可挽救该表型的钙调蛋白抑制剂。我们的研究表明,钙调蛋白抑制剂可降低p53蛋白的量和活性。用钙调蛋白抑制剂治疗导致p53翻译和积累减少,但不影响p53的稳定性。一种美国食品药品监督管理局批准的钙调蛋白抑制剂三氟拉嗪,在斑马鱼和小鼠模型中挽救了DBA模型的造血表型。此外,三氟拉嗪在人CD34造血干细胞和祖细胞中挽救了这些表型。从一名DBA患者分离的CD34细胞中的红系分化也得到改善。这项工作为DBA患者揭示了一条潜在的治疗开发途径。