Blagova Olga, Alieva Indira, Kogan Eugenia, Zaytsev Alexander, Sedov Vsevolod, Chernyavskiy S, Surikova Yulia, Kotov Ilya, Zaklyazminskaya Elena V
Sechenov First Moscow State Medical University, Sechenov University, Moscow, Russia.
Medical Genetics Laboratory, Petrovsky National Research Centre of Surgery, Moscow, Russia.
Front Pharmacol. 2020 Sep 25;11:579450. doi: 10.3389/fphar.2020.579450. eCollection 2020.
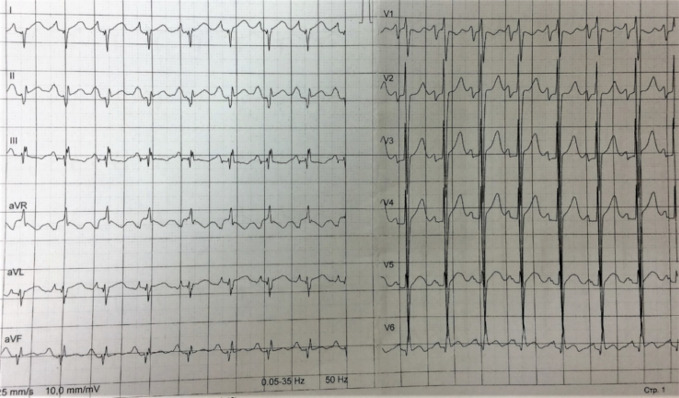
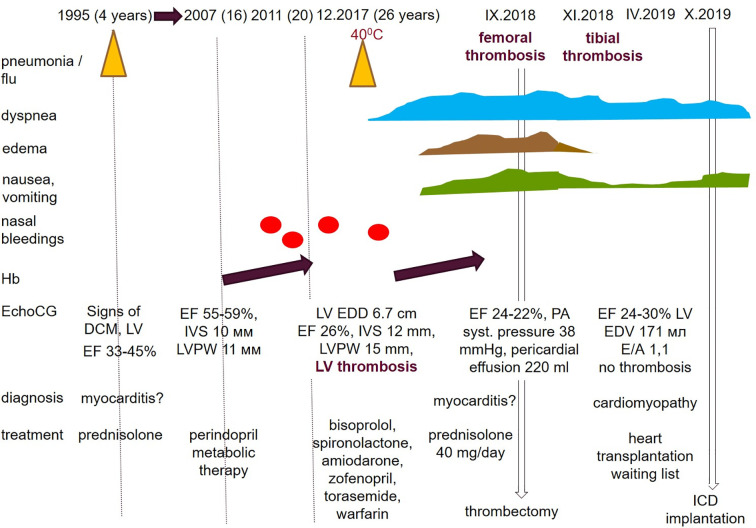
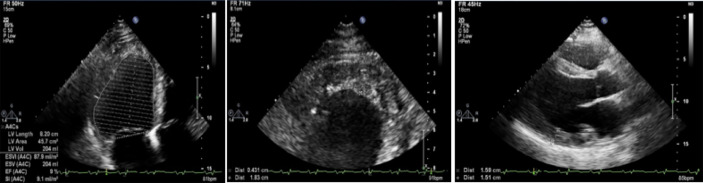
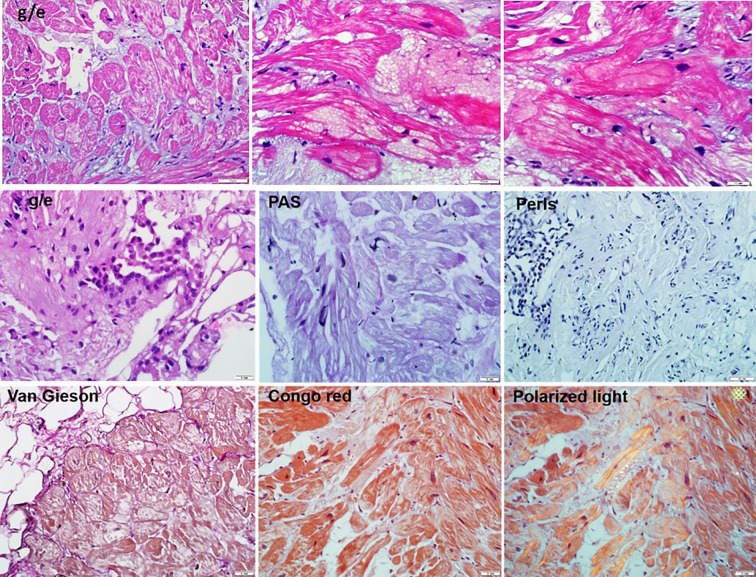
Hypertrophic cardiomyopathy (HCM) is the most common inherited disease, with a prevalence of 1:200 worldwide. The cause of HCM usually presents with an autosomal dominant mutation in the genes encoding one of more than 20 sarcomeric proteins, incomplete penetrance, and variable expressivity. HCM classically manifests as an unexplained thickness of the interventricular septum (IVS) and left ventricular (LV) walls, with or without the obstruction of the LV outflow tract (LVOT), and variable cardiac arrhythmias. Here, we present a rare case of mixed cardiomyopathy (cardiac hypertrophy and dilation) and erythrocytosis in a young patient. A 27-year-old man was admitted to the clinic due to biventricular heart failure (HF) NYHA class III. Personal medical records included a diagnosis of dilated cardiomyopathy (DCM) since the age of 4 years and were, at the time, considered an outcome of myocarditis. Severe respiratory infection led to circulatory decompensation and acute femoral thrombosis. The combination of non-obstructive LV hypertrophy (LV walls up to 15 mm), LV dilatation, decreased contractility (LV EF 24%), and LV apical thrombosis were seen. Cardiac MRI showed a complex pattern of late gadolinium enhancement (LGE). Endomyocardial biopsy (EMB) revealed primary cardiomyopathy with intravascular coagulation and an inflammatory response. No viral genome was detected in the plasma or EMB samples. Whole exome sequencing (WES) revealed a homozygous in-frame deletion p.2711_2737del in the gene. The clinically unaffected mother was a heterozygous carrier of this deletion, and the father was unavailable for clinical and genetic testing. Essential erythrocytosis remains unexplained. No significant improvement was achieved by conventional treatment, including prednisolone 40 mg therapy. ICD was implanted due to sustained VT and high risk of SCD. Orthotopic heart transplantation (HTx) was considered optimal. Early manifestation combined hypertrophic and dilated phenotype, and progression may reflect a complex genotype with more than one pathogenic allele and/or a combination of genetic diseases in one patient.
肥厚型心肌病(HCM)是最常见的遗传性疾病,全球患病率为1:200。HCM的病因通常表现为编码20多种肌节蛋白之一的基因发生常染色体显性突变、不完全外显率和可变表达性。HCM的典型表现为室间隔(IVS)和左心室(LV)壁不明原因增厚,伴或不伴有左心室流出道(LVOT)梗阻以及各种心律失常。在此,我们报告一例年轻患者罕见的混合性心肌病(心脏肥大和扩张)及红细胞增多症病例。一名27岁男性因纽约心脏协会(NYHA)III级双心室心力衰竭(HF)入院。个人病历显示自4岁起诊断为扩张型心肌病(DCM),当时认为是心肌炎的结果。严重呼吸道感染导致循环失代偿和急性股静脉血栓形成。可见非梗阻性左心室肥厚(左心室壁厚度达15毫米)、左心室扩张、收缩力下降(左心室射血分数24%)以及左心室心尖部血栓形成。心脏磁共振成像(MRI)显示钆延迟强化(LGE)的复杂模式。心内膜心肌活检(EMB)显示原发性心肌病伴血管内凝血和炎症反应。血浆或EMB样本中未检测到病毒基因组。全外显子测序(WES)显示该基因存在纯合框内缺失p.2711_2737del。临床未受影响的母亲是该缺失的杂合携带者,父亲无法进行临床和基因检测。真性红细胞增多症的病因仍不明。包括40毫克泼尼松龙治疗在内的传统治疗未取得显著改善。因持续性室性心动过速(VT)和心源性猝死(SCD)高风险植入了植入式心脏复律除颤器(ICD)。原位心脏移植(HTx)被认为是最佳选择。早期表现为肥厚型和扩张型表型并存,病情进展可能反映了一种复杂的基因型,即一名患者存在多个致病等位基因和/或多种遗传疾病的组合。