Institut Hospitalo-Universitaire LIRYC, Pessac.
Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala.
Haematologica. 2021 Feb 1;106(2):337-350. doi: 10.3324/haematol.2020.248153.
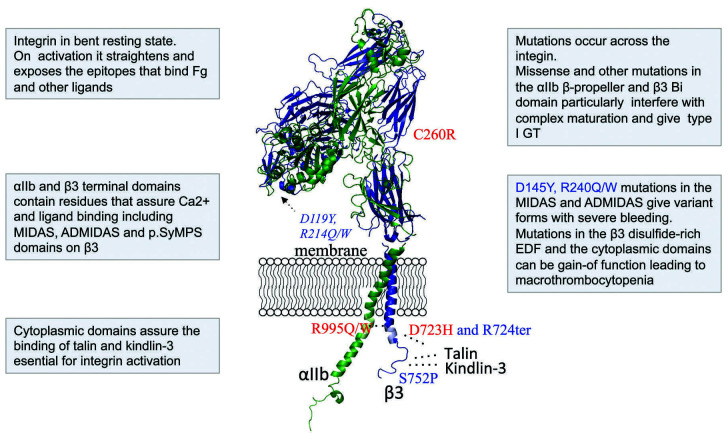
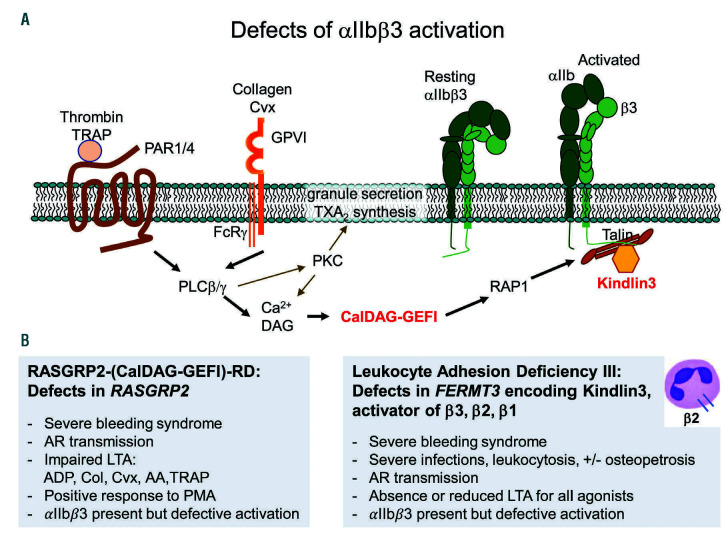
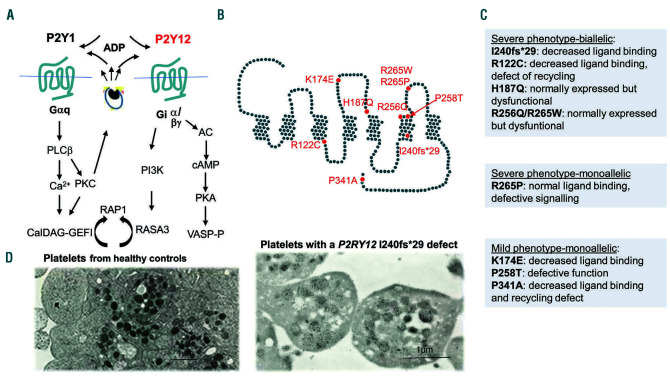
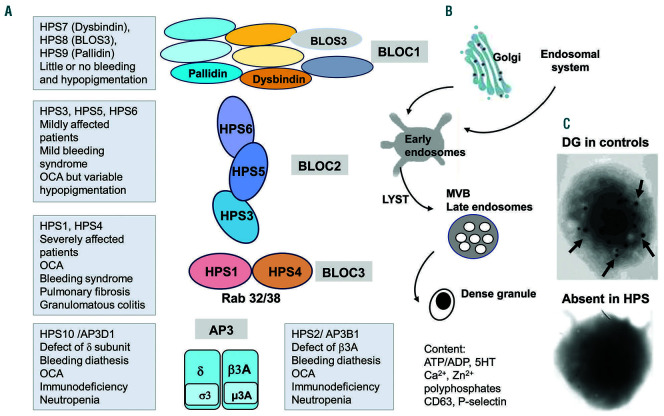
Inherited platelet disorders resulting from platelet function defects and a normal platelet count cause a moderate or severe bleeding diathesis. Since the description of Glanzmann thrombasthenia resulting from defects of ITGA2B and ITGB3, new inherited platelet disorders have been discovered, facilitated by the use of high throughput sequencing and genomic analyses. Defects of RASGRP2 and FERMT3 responsible for severe bleeding syndromes and integrin activation have illustrated the critical role of signaling molecules. Important are mutations of P2RY12 encoding the major ADP receptor causal for an inherited platelet disorder with inheritance characteristics that depend on the variant identified. Interestingly, variants of GP6 encoding the major subunit of the collagen receptor GPVI/FcRγ associate only with mild bleeding. The numbers of genes involved in dense granule defects including Hermansky-Pudlak and Chediak Higashi syndromes continue to progress and are updated. The ANO6 gene encoding a Ca2+-activated ion channel required for phospholipid scrambling is responsible for the rare Scott syndrome and decreased procoagulant activity. A novel EPHB2 defect in a familial bleeding syndrome demonstrates a role for this tyrosine kinase receptor independent of the classical model of its interaction with ephrins. Such advances highlight the large diversity of variants affecting platelet function but not their production, despite the difficulties in establishing a clear phenotype when few families are affected. They have provided insights into essential pathways of platelet function and have been at the origin of new and improved therapies for ischemic disease. Nevertheless, many patients remain without a diagnosis and requiring new strategies that are now discussed.
由于血小板功能缺陷和正常血小板计数导致的遗传性血小板疾病会引起中度或重度出血倾向。自从描述了由 ITGA2B 和 ITGB3 缺陷引起的 Glanzmann 血小板无力症以来,由于高通量测序和基因组分析的应用,已经发现了新的遗传性血小板疾病。RASGRP2 和 FERMT3 的缺陷导致严重的出血综合征和整合素激活,说明了信号分子的关键作用。重要的是,编码主要 ADP 受体的 P2RY12 突变导致一种遗传性血小板疾病,其遗传特征取决于所鉴定的变异。有趣的是,编码胶原受体 GPVI/FcRγ 主要亚基的 GP6 变异仅与轻度出血有关。包括 Hermansky-Pudlak 和 Chediak-Higashi 综合征在内的致密颗粒缺陷相关基因的数量不断增加并得到更新。编码钙激活离子通道的 ANO6 基因是磷脂重排所必需的,它导致罕见的 Scott 综合征和促凝活性降低。家族性出血综合征中新型 EPHB2 缺陷表明,这种酪氨酸激酶受体的作用与其与 ephrins 的经典相互作用模型无关。这些进展突出了影响血小板功能但不影响其产生的大量变异的多样性,尽管在受影响的家庭较少时,建立明确表型存在困难。它们提供了对血小板功能的基本途径的深入了解,并为缺血性疾病的新的和改进的治疗方法奠定了基础。然而,许多患者仍然没有得到诊断,需要新的策略,这些策略现在正在讨论中。