Department of Neurology, Guangdong Province, Guangzhou Women and Children's Medical Center, Guangzhou Medical University, 9# Jin Sui Road, 510623, Guangzhou, People's Republic of China.
BMC Med Genet. 2020 Nov 5;21(1):217. doi: 10.1186/s12881-020-01149-0.
Mitochondrial encephalomyopathy caused by bi-allelic deleterious variants in TARS2 is rare. To date, only two pedigrees were reported in the literature and the connection between the gene and disease needs further study.
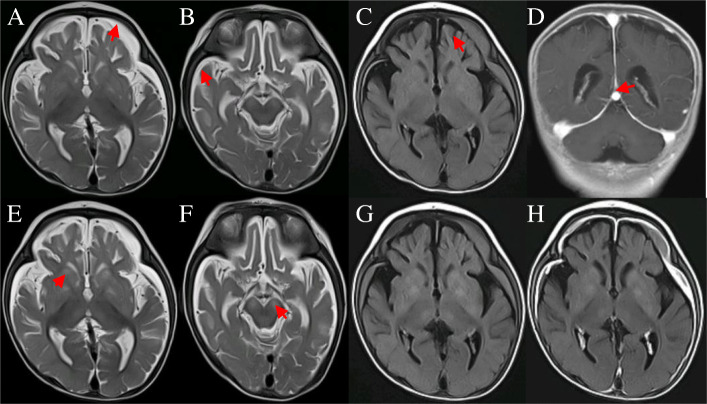
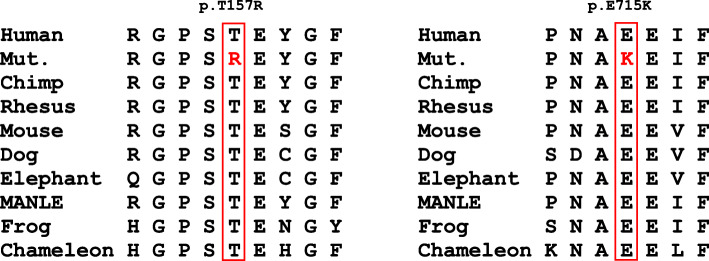
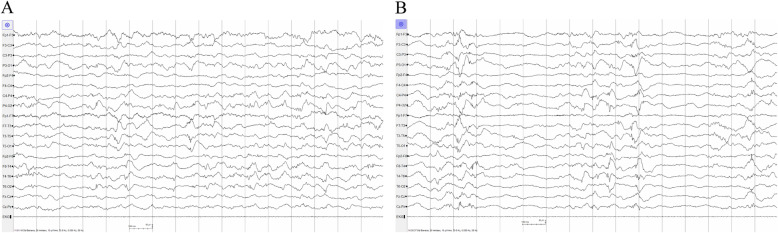
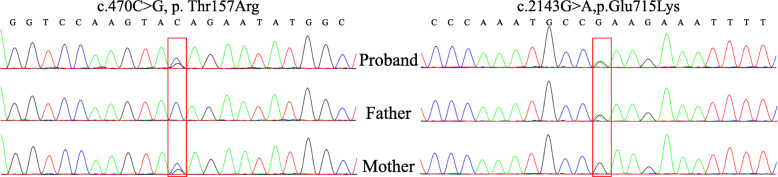
We report one infant who presented with limb hypertonia, epilepsy, developmental delay, and increased serum lactate from a non-consanguineous Chinese family. Whole-genome sequencing was performed to help to underlie the cause. We identified compound heterozygous variants c.470C > G, p.Thr157Arg and c.2143G > A, p.Glu715Lys in TARS2 and the variants were confirmed by Sanger sequencing. The patient was diagnosed with combined oxidative phosphorylation deficiency 21 according to the Online Mendelian Inheritance in Man (OMIM) database based on the clinical data and the deleterious effect of the two variants in TARS2 predicted by in silico tools.
We presented one case diagnosed with combined oxidative phosphorylation deficiency 21 based on clinical characteristics and genetic analysis. This is the first case in China and the fourth case in the world based on our document retrieval. This study facilitates the understanding of combined oxidative phosphorylation deficiency disease and demonstrates that the next-generation sequencing has a high potential to study inherited disease with high phenotypic heterogeneity and genetic heterogeneity including mitochondrial diseases such as combined oxidative phosphorylation deficiency.
由 TARS2 双等位有害变异引起的线粒体脑肌病很少见。迄今为止,文献中仅报道了两大家系,该基因与疾病之间的联系仍需要进一步研究。
我们报告了一例来自非近亲中国家庭的婴儿,其表现为肢体痉挛、癫痫、发育迟缓以及血清乳酸升高。进行全基因组测序以帮助阐明病因。我们在 TARS2 中发现了复合杂合变异 c.470C>G,p.Thr157Arg 和 c.2143G>A,p.Glu715Lys,并通过 Sanger 测序进行了确认。根据在线孟德尔遗传在线数据库(OMIM),根据临床数据和 TARS2 中两个变异的致病变异预测工具,该患者被诊断为联合氧化磷酸化缺陷 21。
我们根据临床特征和基因分析诊断了一例联合氧化磷酸化缺陷 21。这是基于我们的文献检索,在中国的首例病例,也是全球的第 4 例病例。该研究有助于理解联合氧化磷酸化缺陷疾病,并表明下一代测序具有研究包括线粒体疾病在内的具有高度表型异质性和遗传异质性的遗传疾病的巨大潜力。