Silva Diana F, Candeias Emanuel, Esteves A Raquel, Magalhães João D, Ferreira I Luísa, Nunes-Costa Daniela, Rego A Cristina, Empadinhas Nuno, Cardoso Sandra M
CNC-Center for Neuroscience and Cell Biology, University of Coimbra, Largo Marquês de Pombal, 3004-517, Coimbra, Portugal.
IIIUC-Institute for Interdisciplinary Research, University of Coimbra, Coimbra, Portugal.
J Neuroinflammation. 2020 Nov 5;17(1):332. doi: 10.1186/s12974-020-02004-y.
After decades of research recognizing it as a complex multifactorial disorder, sporadic Alzheimer's disease (sAD) still has no known etiology. Adding to the myriad of different pathways involved, bacterial neurotoxins are assuming greater importance in the etiology and/or progression of sAD. β-N-Methylamino-L-alanine (BMAA), a neurotoxin produced by some microorganisms namely cyanobacteria, was previously detected in the brains of AD patients. Indeed, the consumption of BMAA-enriched foods has been proposed to induce amyotrophic lateral sclerosis-parkinsonism-dementia complex (ALS-PDC), which implicated this microbial metabolite in neurodegeneration mechanisms.
Freshly isolated mitochondria from C57BL/6 mice were treated with BMAA and O consumption rates were determined. O consumption and glycolysis rates were also measured in mouse primary cortical neuronal cultures. Further, mitochondrial membrane potential and ROS production were evaluated by fluorimetry and the integrity of mitochondrial network was examined by immunofluorescence. Finally, the ability of BMAA to activate neuronal innate immunity was quantified by addressing TLRs (Toll-like receptors) expression, p65 NF-κB translocation into the nucleus, increased expression of NLRP3 (Nod-like receptor 3), and pro-IL-1β. Caspase-1 activity was evaluated using a colorimetric substrate and mature IL-1β levels were also determined by ELISA.
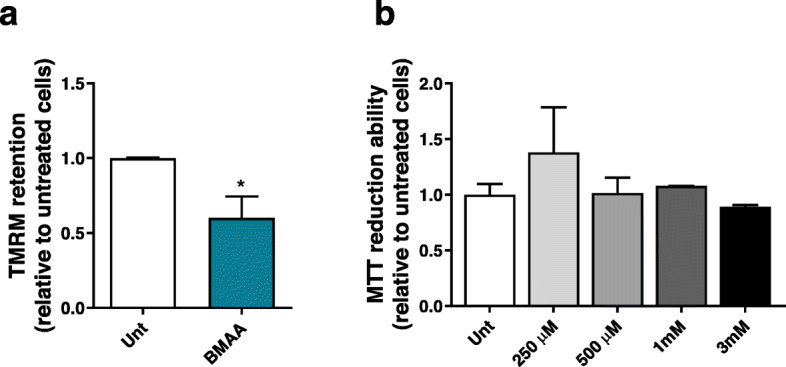
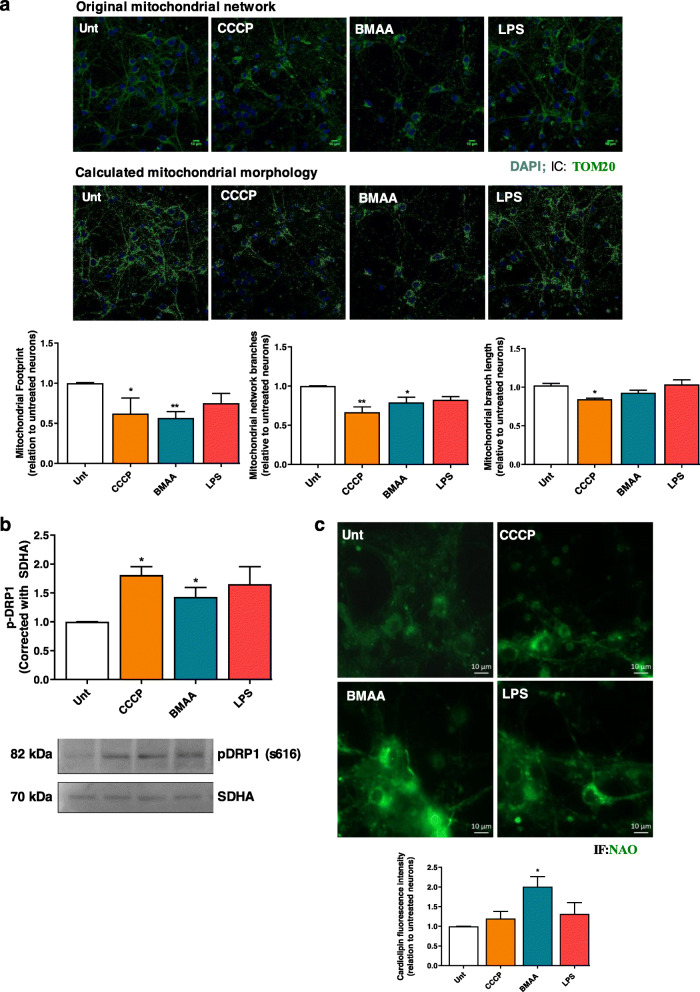
Treatment with BMAA reduced O consumption rates in both isolated mitochondria and in primary cortical cultures, with additional reduced glycolytic rates, decrease mitochondrial potential and increased ROS production. The mitochondrial network was found to be fragmented, which resulted in cardiolipin exposure that stimulated inflammasome NLRP3, reinforced by decreased mitochondrial turnover, as indicated by increased p62 levels. BMAA treatment also activated neuronal extracellular TLR4 and intracellular TLR3, inducing p65 NF-κB translocation into the nucleus and activating the transcription of NLRP3 and pro-IL-1β. Increased caspase-1 activity resulted in elevated levels of mature IL-1β. These alterations in mitochondrial metabolism and inflammation increased Tau phosphorylation and Aβ peptides production, two hallmarks of AD.
Here we propose a unifying mechanism for AD neurodegeneration in which a microbial toxin can induce mitochondrial dysfunction and activate neuronal innate immunity, which ultimately results in Tau and Aβ pathology. Our data show that neurons, alone, can mount inflammatory responses, a role previously attributed exclusively to glial cells.
经过数十年将散发性阿尔茨海默病(sAD)认定为一种复杂的多因素疾病的研究,其病因仍不明。在众多涉及的不同途径中,细菌神经毒素在sAD的病因和/或进展中变得愈发重要。β - N - 甲基氨基 - L - 丙氨酸(BMAA)是一种由某些微生物(即蓝藻)产生的神经毒素,此前在AD患者的大脑中被检测到。事实上,有人提出食用富含BMAA的食物会诱发肌萎缩侧索硬化 - 帕金森病 - 痴呆综合征(ALS - PDC),这表明这种微生物代谢产物参与了神经退行性变机制。
用BMAA处理从C57BL/6小鼠新鲜分离的线粒体,并测定氧气消耗率。还在小鼠原代皮质神经元培养物中测量氧气消耗和糖酵解率。此外,通过荧光测定法评估线粒体膜电位和活性氧(ROS)的产生,并通过免疫荧光检查线粒体网络的完整性。最后,通过检测Toll样受体(TLRs)表达、p65核因子κB(NF - κB)转位到细胞核、Nod样受体3(NLRP3)表达增加以及白细胞介素 - 1β前体(pro - IL - 1β)来量化BMAA激活神经元固有免疫的能力。使用比色底物评估半胱天冬酶 - 1的活性,并通过酶联免疫吸附测定(ELISA)确定成熟白细胞介素 - 1β的水平。
用BMAA处理可降低分离线粒体和原代皮质培养物中的氧气消耗率,同时糖酵解率进一步降低,线粒体电位降低,ROS产生增加。发现线粒体网络碎片化,导致心磷脂暴露,刺激了炎性小体NLRP3,p62水平升高表明线粒体更新减少进一步加剧了这种情况。BMAA处理还激活了神经元细胞外的TLR4和细胞内的TLR3,诱导p65 NF - κB转位到细胞核并激活NLRP3和pro - IL - 1β的转录。半胱天冬酶 - 1活性增加导致成熟白细胞介素 - 1β水平升高。线粒体代谢和炎症的这些改变增加了Tau蛋白磷酸化和β淀粉样肽(Aβ)的产生,这是AD的两个特征。
在此我们提出一种AD神经退行性变的统一机制,即一种微生物毒素可诱导线粒体功能障碍并激活神经元固有免疫,最终导致Tau和Aβ病理改变。我们的数据表明,神经元自身就能引发炎症反应,而这一作用以前仅被认为是神经胶质细胞所特有的。