Unit of Animal Genomics, GIGA-R & Faculty of Veterinary Medicine, University of Liège, Liège, Belgium.
Centro de Investigación Mariña, Departamento de Bioquímica, Genética e Inmunología, Edificio CC Experimentais, Universidade de Vigo, Campus de Vigo, As Lagoas, Marcosende, 36310, Vigo, Spain.
Heredity (Edinb). 2021 Mar;126(3):410-423. doi: 10.1038/s41437-020-00383-9. Epub 2020 Nov 6.
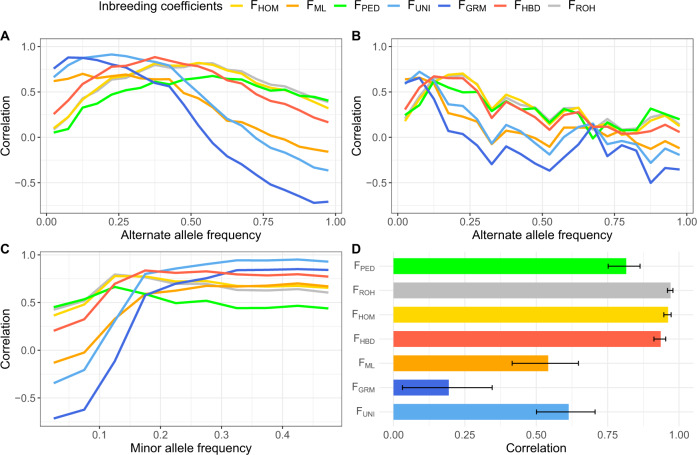
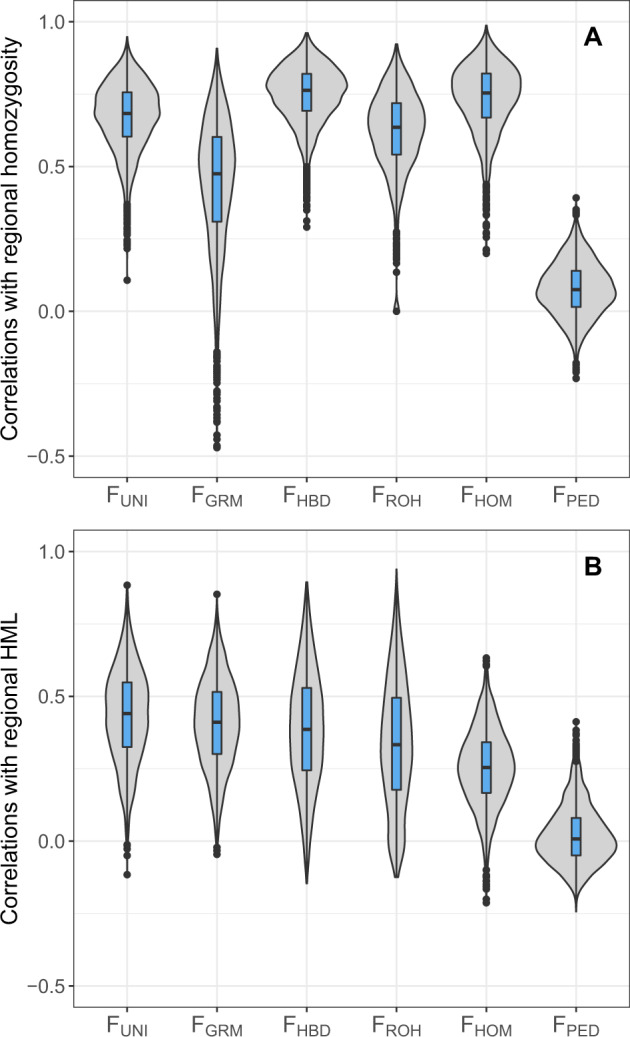
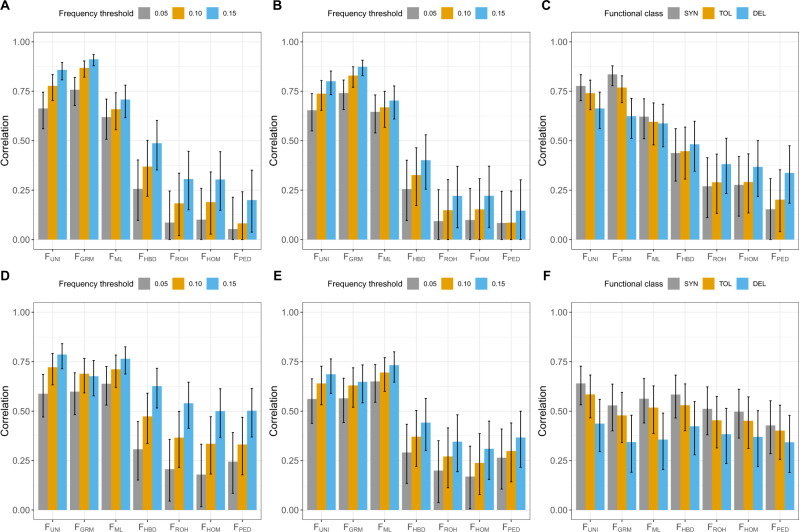
The estimation of the inbreeding coefficient (F) is essential for the study of inbreeding depression (ID) or for the management of populations under conservation. Several methods have been proposed to estimate the realized F using genetic markers, but it remains unclear which one should be used. Here we used whole-genome sequence data for 245 individuals from a Holstein cattle pedigree to empirically evaluate which estimators best capture homozygosity at variants causing ID, such as rare deleterious alleles or loci presenting heterozygote advantage and segregating at intermediate frequency. Estimators relying on the correlation between uniting gametes (F) or on the genomic relationships (F) presented the highest correlations with these variants. However, homozygosity at rare alleles remained poorly captured. A second group of estimators relying on excess homozygosity (F), homozygous-by-descent segments (F), runs-of-homozygosity (F) or on the known genealogy (F) was better at capturing whole-genome homozygosity, reflecting the consequences of inbreeding on all variants, and for young alleles with low to moderate frequencies (0.10 < . < 0.25). The results indicate that F and F might present a stronger association with ID. However, the situation might be different when recessive deleterious alleles reach higher frequencies, such as in populations with a small effective population size. For locus-specific inbreeding measures or at low marker density, the ranking of the methods can also change as F makes better use of the information from neighboring markers. Finally, we confirmed that genomic measures are in general superior to pedigree-based estimates. In particular, F was uncorrelated with locus-specific homozygosity.
近亲系数 (F) 的估计对于研究近交衰退 (ID) 或管理保护下的种群至关重要。已经提出了几种使用遗传标记估计实际 F 的方法,但仍不清楚应该使用哪种方法。在这里,我们使用了来自荷斯坦奶牛系谱的 245 个个体的全基因组序列数据,从经验上评估了哪些估算器最能捕捉导致 ID 的变异的同质性,例如罕见的有害等位基因或表现出杂合优势且在中间频率下分离的基因座。依赖于联合配子之间相关性 (F) 或基于基因组关系 (F) 的估算器与这些变异的相关性最高。然而,罕见等位基因的同质性仍然难以捕捉。第二类依赖于过剩同质性 (F)、同源单倍型 (F)、纯合片段 (F)、纯合段长度 (F) 或已知系谱 (F) 的估算器更能捕捉全基因组同质性,反映了近交对所有变异的影响,并且对于低频到中度频率 (0.10 <. < 0.25) 的年轻等位基因也是如此。结果表明,F 和 F 可能与 ID 具有更强的关联。然而,当隐性有害等位基因达到更高频率时,情况可能会有所不同,例如在有效种群大小较小的群体中。对于特定基因座的近交度量或在低标记密度下,方法的排名也可能会发生变化,因为 F 可以更好地利用来自邻近标记的信息。最后,我们证实了基因组度量通常优于基于系谱的估计。特别是,F 与特定基因座的同质性不相关。