Saha Otun, Hossain Md Shahadat, Rahaman Md Mizanur
Department of Microbiology, University of Dhaka, Dhaka 1000, Bangladesh.
Department of Biotechnology and Genetic Engineering, Noakhali Science and Technology University, Noakhali 3814, Bangladesh.
Gene Rep. 2020 Dec;21:100951. doi: 10.1016/j.genrep.2020.100951. Epub 2020 Nov 1.
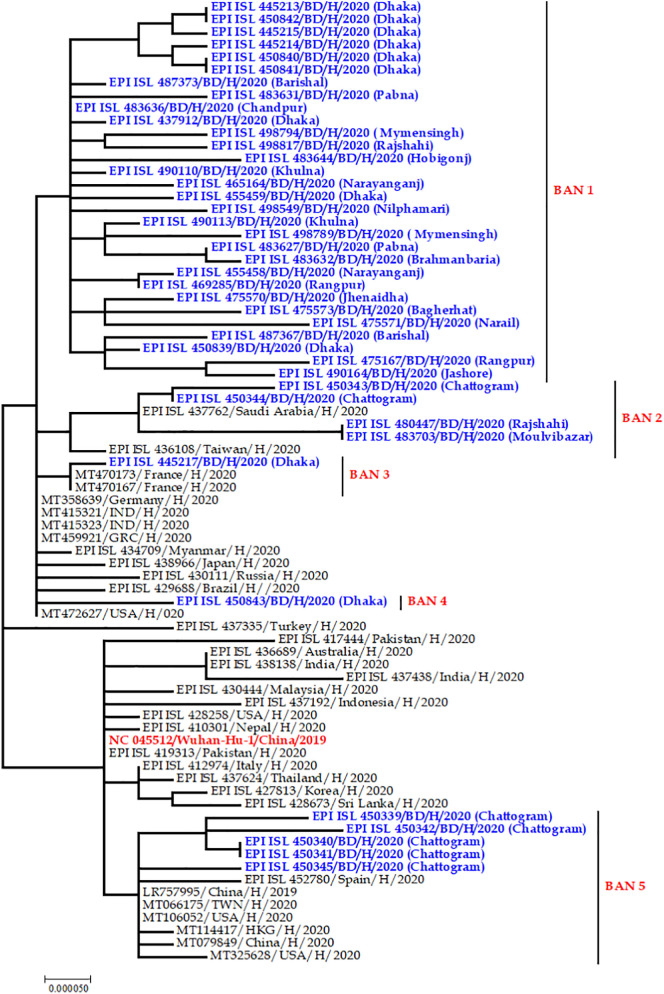
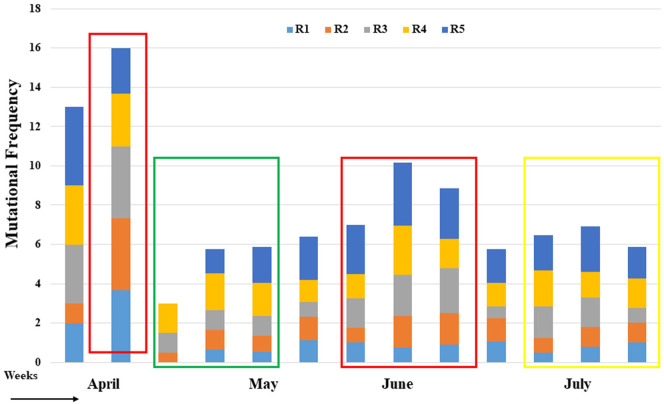
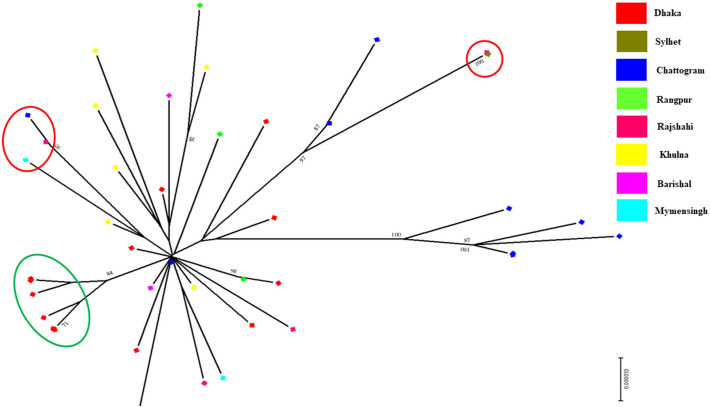
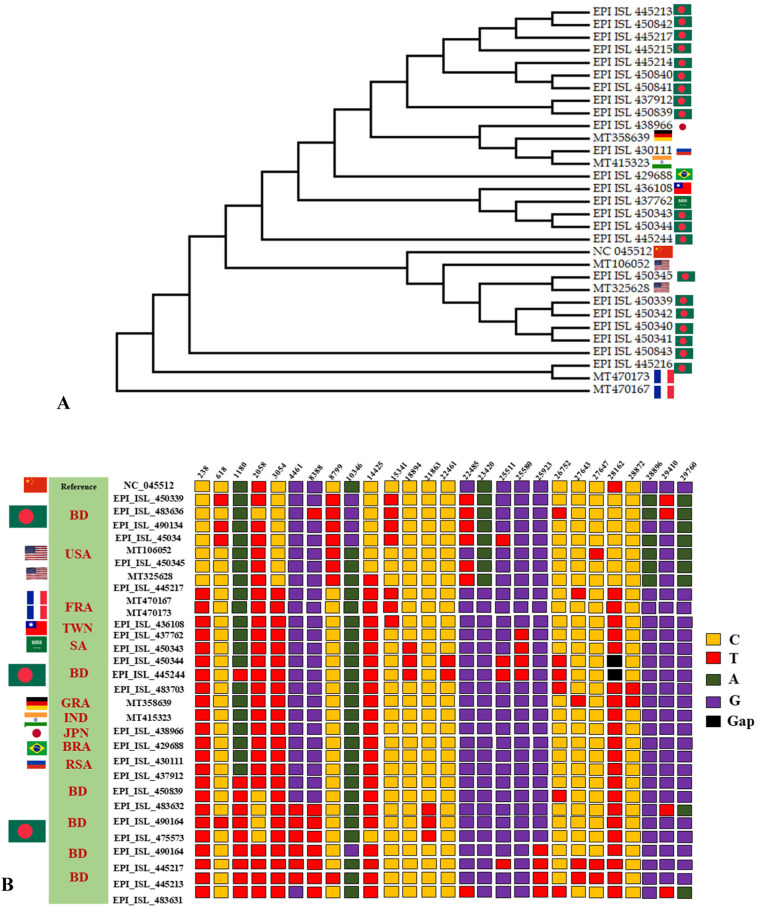
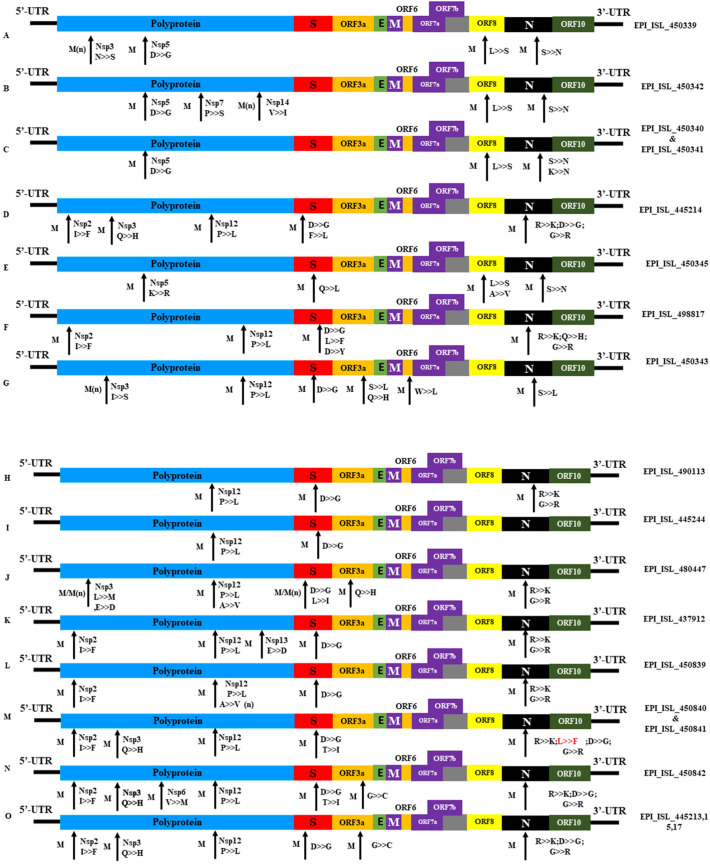
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a new strain of beta coronavirus that has spread worldwide within a short period of time and has been responsible for the current COVID-19 pandemic. This novel virus shows high transmission and adaptability frequency into the host with rapid changes in genomic sequences. In this study, we analyzed the complete genome of 41 strains isolated in Bangladesh to understand the evolutionary route and genetic variations of this rapidly evolving virus. The phylogenetics, parsimony informative sites and mutation analyses were performed using MEGA X, Multiple sequence alignment program (MAFFT), and Virus Pathogen Resource. The phylogenetic analysis of the studied genomes along with the reference genome suggested that the viral strains found in Bangladesh might be coming from multiple countries such as France, Germany, India, the USA, and Brazil. After entering into the country, intra-cluster and inter-cluster began to circulate in the 8 individual divisions of Bangladesh. We also identified 26 parsimony-informative sites along with the 9 most important sites for virus evolution. Genome-wide annotations revealed 256 mutations, of which 10 were novel (NSP3, RdRp, Spike) in Bangladeshi strains where I120F(NSP2), P323L(RdRp), D614G (Spike), R203K, G204R(N) are the most prominent. Most importantly, numerous mutations were flourishing in the N protein gene (67) followed by S (45), RdRp (38), NSP2 (34), NSP3 (20), and ORF8 (6) gene. Moreover, nucleotide deletion analysis found nine deletions throughout the genomes including in ORF7a (8), ORF8 (1) with one insertion (G) at 265 positions in only one genome. The underlying mechanism of disease severity, molecular evolution, and epidemiology lie in genomic sequences that are not fully understood yet. Identification of the evolutionary history, parsimony-informative sites and others genetic variations of this deadly virus will facilitate the development of new strategies to control the local transmission and provide deep insight in the identification of potential therapeutic targets for controlling COVID-19.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)是一种新型的β冠状病毒,在短时间内已在全球范围内传播,并导致了当前的COVID-19大流行。这种新型病毒显示出高传播性和对宿主的适应性,其基因组序列变化迅速。在本研究中,我们分析了在孟加拉国分离出的41株病毒的完整基因组,以了解这种快速进化的病毒的进化路径和基因变异。使用MEGA X、多序列比对程序(MAFFT)和病毒病原体资源进行了系统发育分析、简约信息位点分析和突变分析。对所研究的基因组与参考基因组进行的系统发育分析表明,在孟加拉国发现的病毒株可能来自多个国家,如法国、德国、印度、美国和巴西。进入该国后,病毒在孟加拉国的8个单独行政区内开始在簇内和簇间传播。我们还确定了26个简约信息位点以及9个对病毒进化最重要的位点。全基因组注释揭示了256个突变,其中10个是孟加拉国病毒株中的新突变(NSP3、RdRp、刺突蛋白),其中I120F(NSP2)、P323L(RdRp)、D614G(刺突蛋白)、R203K、G204R(N)最为突出。最重要的是,N蛋白基因中出现了大量突变(67个),其次是S(45个)、RdRp(38个)、NSP2(34个)、NSP3(20个)和ORF8(6个)基因。此外,核苷酸缺失分析在整个基因组中发现了9处缺失,包括ORF7a(8处)、ORF8(1处),仅在一个基因组的265位有一处插入(G)。疾病严重程度、分子进化和流行病学的潜在机制在于尚未完全了解的基因组序列。确定这种致命病毒的进化历史、简约信息位点和其他基因变异将有助于制定控制本地传播的新策略,并为确定控制COVID-19的潜在治疗靶点提供深入见解。