Dinu Dennis F, Podewitz Maren, Grothe Hinrich, Loerting Thomas, Liedl Klaus R
Institute of General, Inorganic and Theoretical Chemistry, University of Innsbruck, Innsbruck, Austria.
Institute of Material Chemistry, TU Vienna, Vienna, Austria.
Theor Chem Acc. 2020;139(12):174. doi: 10.1007/s00214-020-02682-0. Epub 2020 Nov 9.
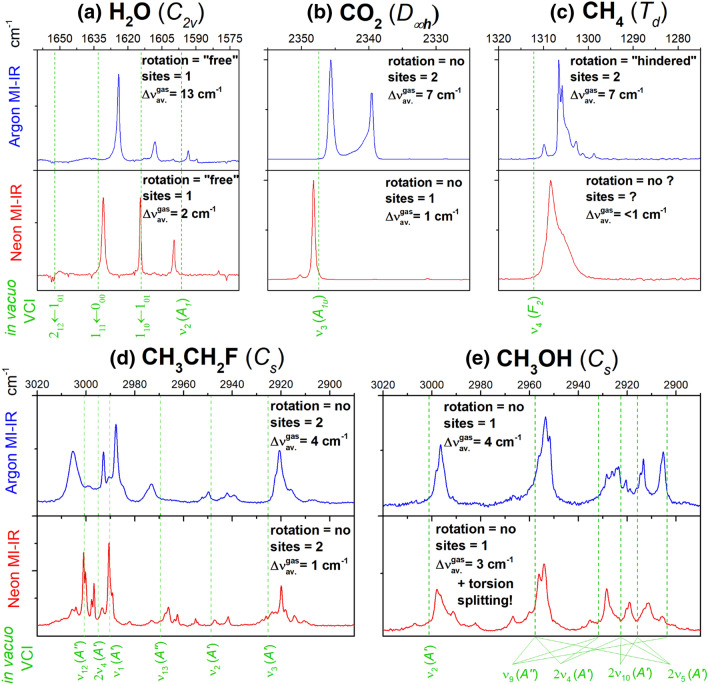
The key feature of matrix-isolation infrared (MI-IR) spectroscopy is the isolation of single guest molecules in a host system at cryogenic conditions. The matrix mostly hinders rotation of the guest molecule, providing access to pure vibrational features. Vibrational self-consistent field (VSCF) and configuration interaction computations (VCI) on ab initio multimode potential energy surfaces (PES) give rise to anharmonic vibrational spectra. In a single-sourced combination of these experimental and computational approaches, we have established an iterative spectroscopic characterization procedure. The present article reviews the scope of this procedure by highlighting the strengths and limitations based on the examples of water, carbon dioxide, methane, methanol, and fluoroethane. An assessment of setups for the construction of the multimode PES on the example of methanol demonstrates that CCSD(T)-F12 level of theory is preferable to compute (a) accurate vibrational frequencies and (b) equilibrium or vibrationally averaged structural parameters. Our procedure has allowed us to uniquely assign unknown or disputed bands and enabled us to clarify problematic spectral regions that are crowded with combination bands and overtones. Besides spectroscopic assignment, the excellent agreement between theory and experiment paves the way to tackle questions of rather fundamental nature as to whether or not matrix effects are systematic, and it shows the limits of conventional notations used by spectroscopists.
基质隔离红外(MI-IR)光谱的关键特性是在低温条件下将单个客体分子隔离在主体体系中。基质大多会阻碍客体分子的转动,从而获得纯净的振动特征。基于从头算多模势能面(PES)的振动自洽场(VSCF)和组态相互作用计算(VCI)可产生非谐振动光谱。通过将这些实验和计算方法进行单一来源的结合,我们建立了一种迭代光谱表征程序。本文以水、二氧化碳、甲烷、甲醇和氟乙烷为例,通过强调其优势和局限性来回顾该程序的适用范围。以甲醇为例对构建多模PES的装置进行的评估表明,CCSD(T)-F12理论水平在计算(a)精确的振动频率和(b)平衡或振动平均结构参数方面更具优势。我们的程序使我们能够唯一地归属未知或有争议的谱带,并使我们能够厘清那些被组合带和泛音所充斥的有问题的光谱区域。除了光谱归属外,理论与实验之间的高度吻合为解决诸如基质效应是否具有系统性等相当基础的问题铺平了道路,并且它还展示了光谱学家所使用的传统符号的局限性。