Krasnov George S, Pushkova Elena N, Novakovskiy Roman O, Kudryavtseva Ludmila P, Rozhmina Tatiana A, Dvorianinova Ekaterina M, Povkhova Liubov V, Kudryavtseva Anna V, Dmitriev Alexey A, Melnikova Nataliya V
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, Russia.
Federal Research Center for Bast Fiber Crops, Torzhok, Russia.
Front Genet. 2020 Aug 27;11:959. doi: 10.3389/fgene.2020.00959. eCollection 2020.
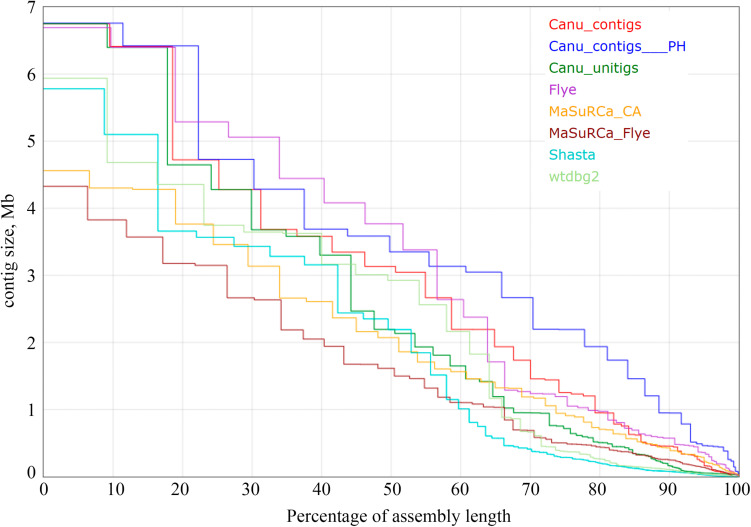
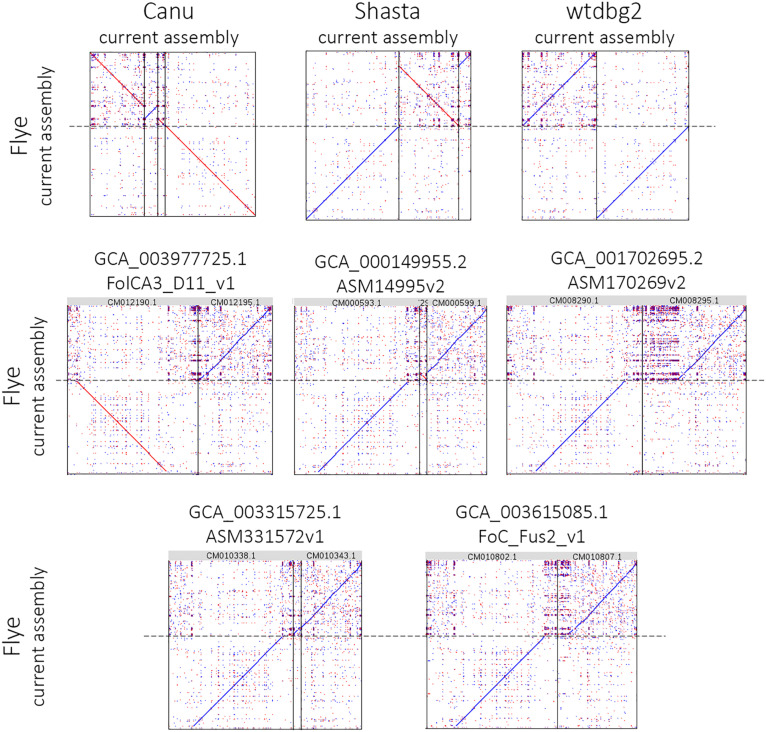
In the present work, a highly pathogenic isolate of f. sp. , which is the most harmful pathogen affecting flax ( L.), was sequenced for the first time. To achieve a high-quality genome assembly, we used the combination of two sequencing platforms - Oxford Nanopore Technologies (MinION system) with long noisy reads and Illumina (HiSeq 2500 instrument) with short accurate reads. Given the quality of DNA is crucial for Nanopore sequencing, we developed the protocol for extraction of pure high-molecular-weight DNA from fungi. Sequencing of DNA extracted using this protocol allowed us to obtain about 85x genome coverage with long (N50 = 29 kb) MinION reads and 30x coverage with 2 × 250 bp HiSeq reads. Several tools were developed for genome assembly; however, they provide different results depending on genome complexity, sequencing data volume, read length and quality. We benchmarked the most requested assemblers (Canu, Flye, Shasta, wtdbg2, and MaSuRCA), Nanopore polishers (Medaka and Racon), and Illumina polishers (Pilon and POLCA) on our sequencing data. The assembly performed with Canu and polished with Medaka and POLCA was considered the most full and accurate. After further elimination of redundant contigs using Purge Haplotigs, we achieved a high-quality genome of f. sp. with a total length of 59 Mb, N50 of 3.3 Mb, and 99.5% completeness according to BUSCO. We also obtained a complete circular mitochondrial genome with a length of 38.7 kb. The achieved assembly expands studies on and plant-pathogen interaction in flax.
在本研究中,首次对一种高致病性的f. sp.分离株进行了测序,该分离株是影响亚麻(L.)的最有害病原体。为了实现高质量的基因组组装,我们使用了两种测序平台的组合——具有长且有噪声读段的牛津纳米孔技术(MinION系统)和具有短且准确读段的Illumina(HiSeq 2500仪器)。鉴于DNA质量对于纳米孔测序至关重要,我们开发了从真菌中提取纯高分子量DNA的方案。使用该方案提取的DNA测序使我们能够获得约85倍基因组覆盖度的长MinION读段(N50 = 29 kb)和30倍覆盖度的2×250 bp HiSeq读段。开发了几种用于基因组组装的工具;然而,它们根据基因组复杂性、测序数据量、读段长度和质量提供不同的结果。我们在我们的测序数据上对最常用的组装器(Canu、Flye、Shasta、wtdbg2和MaSuRCA)、纳米孔抛光器(Medaka和Racon)以及Illumina抛光器(Pilon和POLCA)进行了基准测试。使用Canu进行组装并使用Medaka和POLCA进行抛光被认为是最完整和准确的。在使用Purge Haplotigs进一步消除冗余重叠群后,我们获得了一个高质量的f. sp.基因组,总长度为59 Mb,N50为3.3 Mb,根据BUSCO评估的完整性为99.5%。我们还获得了一个长度为38.7 kb的完整环状线粒体基因组。所实现的组装扩展了对亚麻中该病原体与植物病原体相互作用的研究。