LAQV-REQUIMTE, Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, 4169-007 Porto, Portugal.
Soft Matter and Molecular Biophysics Group, Department of Applied Physics, University of Santiago de Compostela, 15782 Santiago de Compostela, Spain.
Molecules. 2020 Nov 19;25(22):5425. doi: 10.3390/molecules25225425.
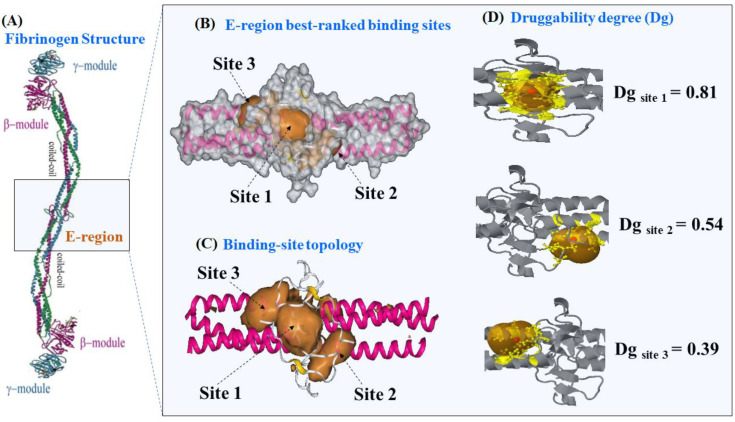
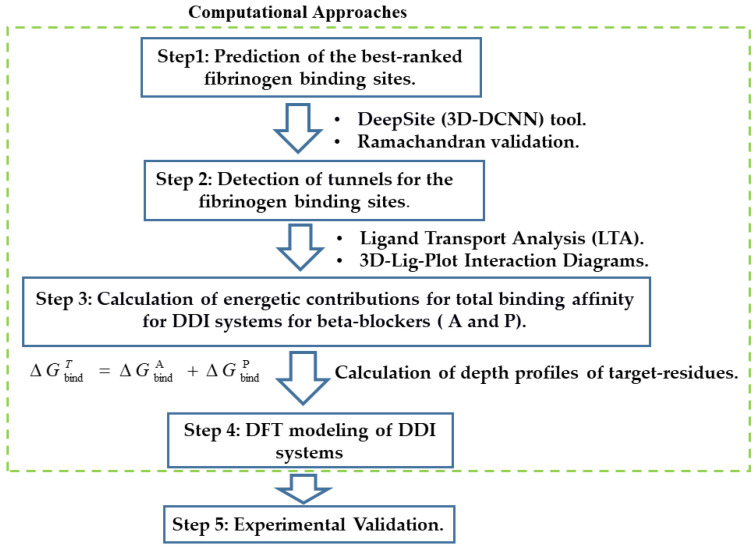
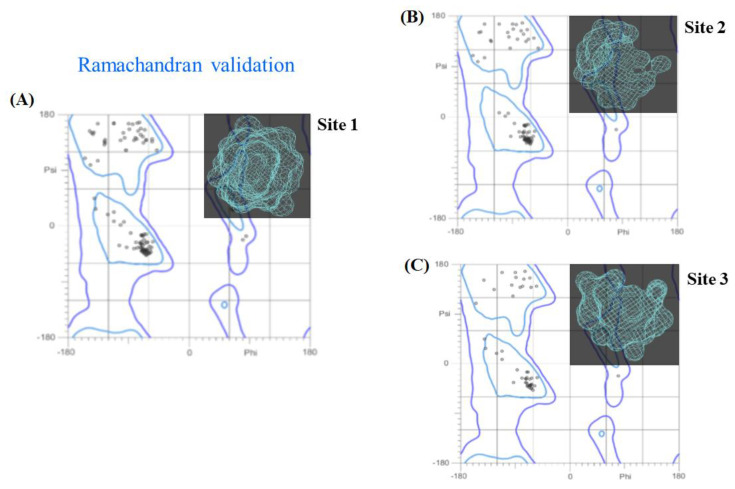
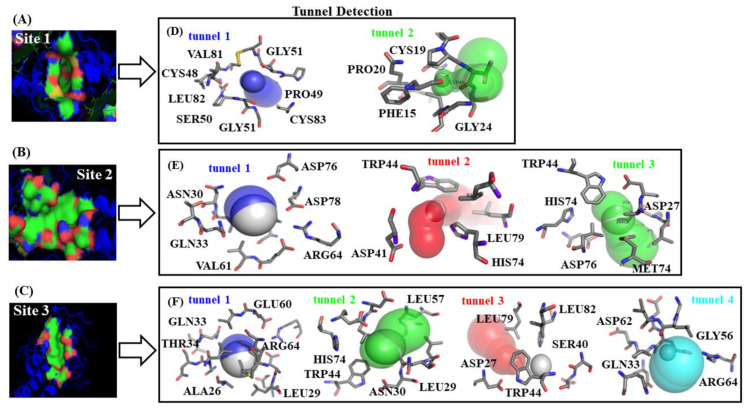
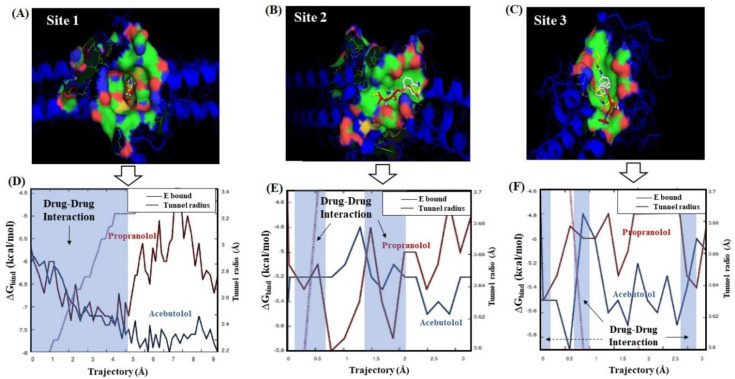
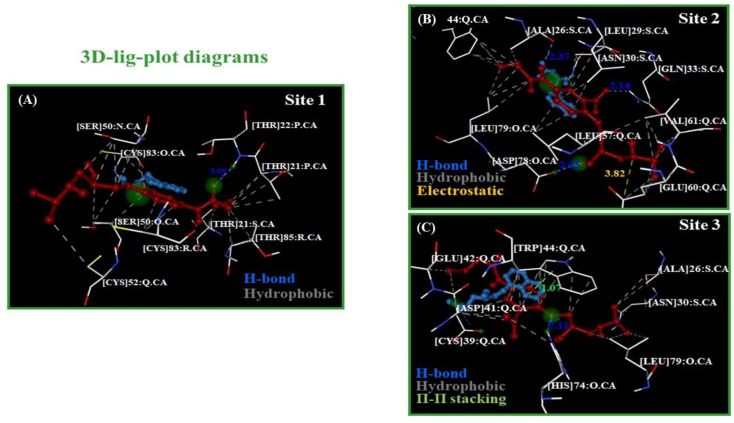
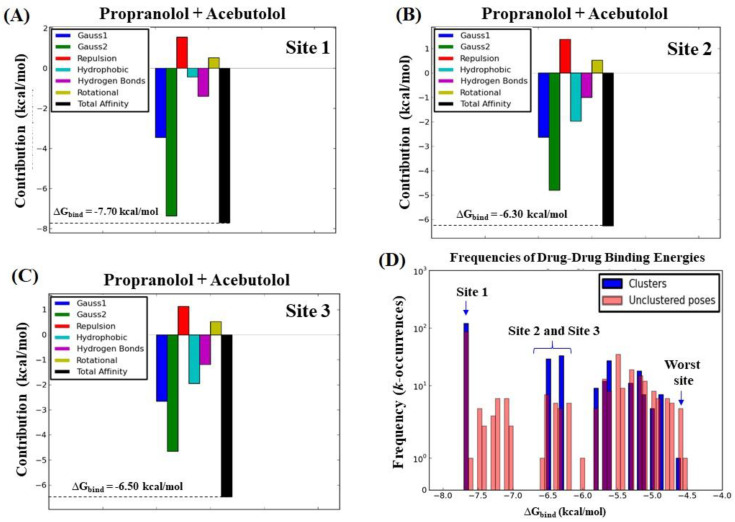
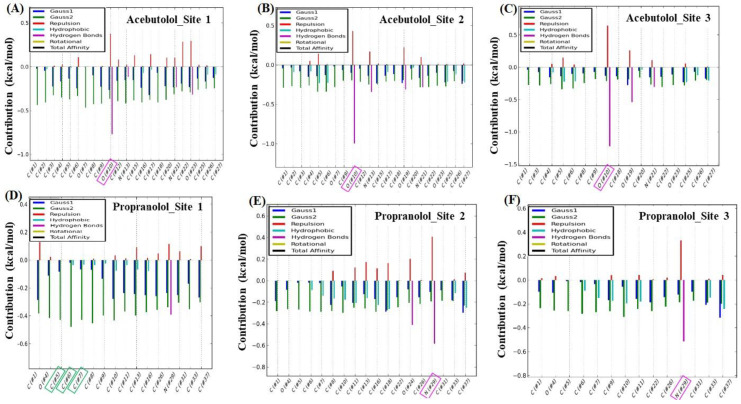
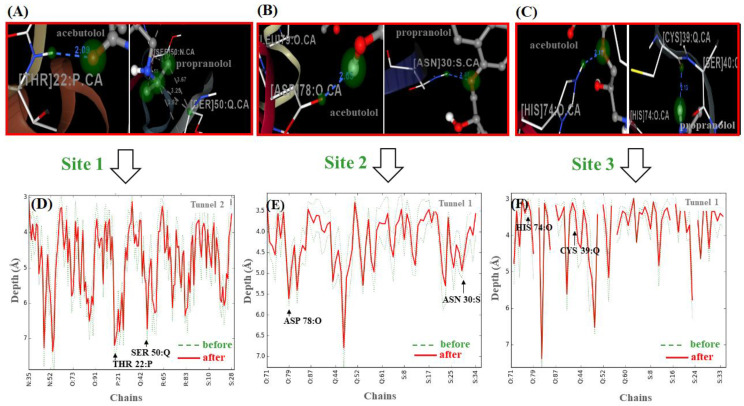
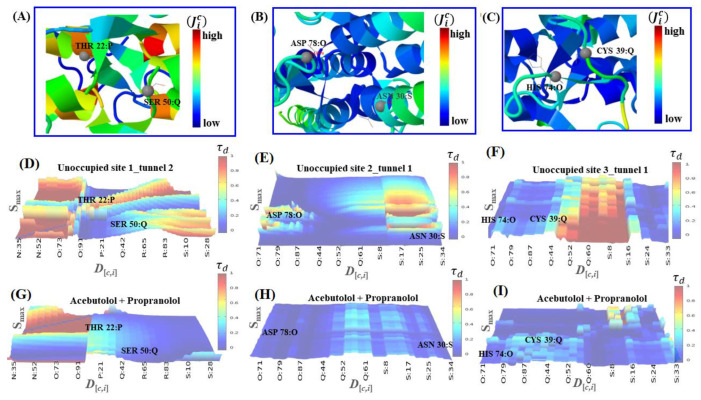
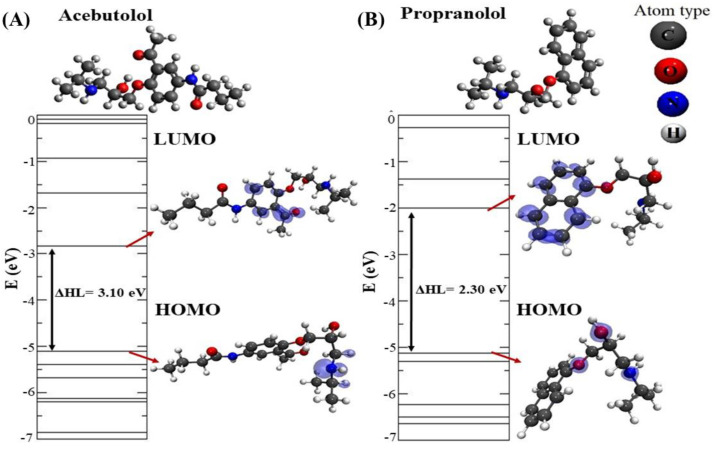
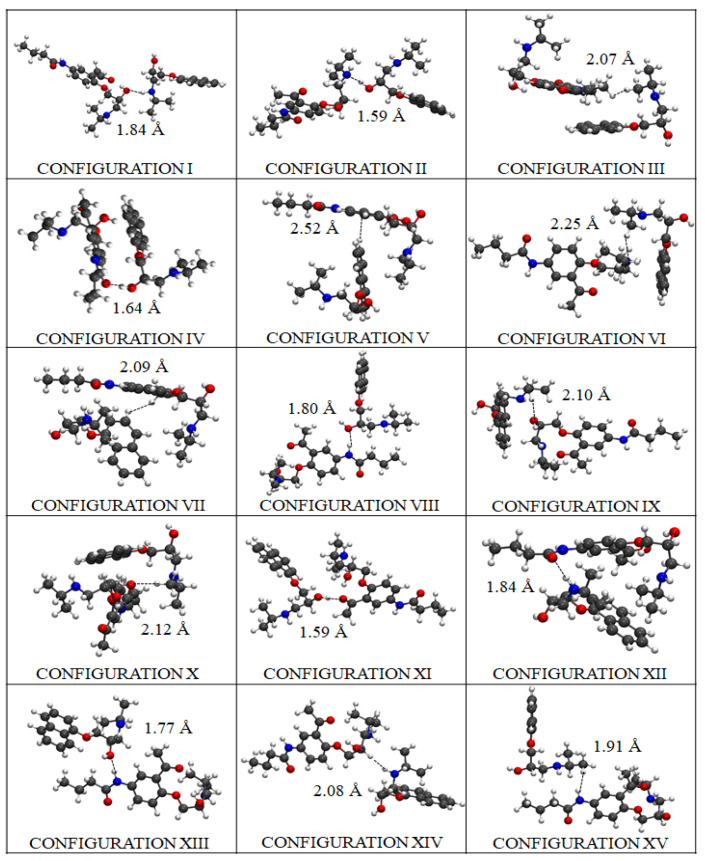
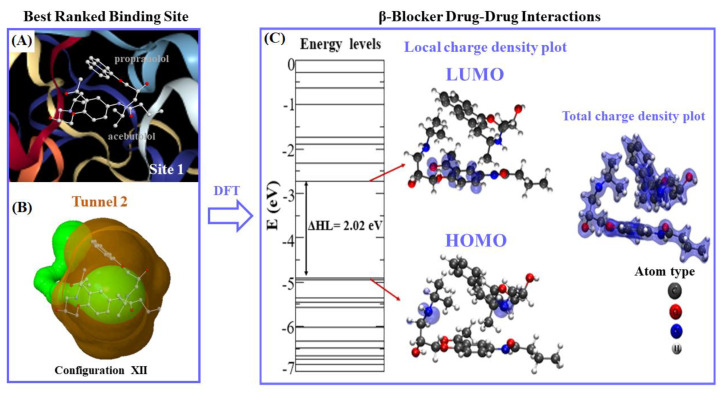
In this work, one of the most prevalent polypharmacology drug-drug interaction events that occurs between two widely used beta-blocker drugs-i.e., acebutolol and propranolol-with the most abundant blood plasma fibrinogen protein was evaluated. Towards that end, molecular docking and Density Functional Theory (DFT) calculations were used as complementary tools. A fibrinogen crystallographic validation for the three best ranked binding-sites shows 100% of conformationally favored residues with total absence of restricted flexibility. From those three sites, results on both the binding-site druggability and ligand transport analysis-based free energy trajectories pointed out the most preferred biophysical environment site for drug-drug interactions. Furthermore, the total affinity for the stabilization of the drug-drug complexes was mostly influenced by steric energy contributions, based mainly on multiple hydrophobic contacts with critical residues (THR22: P and SER50: Q) in such best-ranked site. Additionally, the DFT calculations revealed that the beta-blocker drug-drug complexes have a spontaneous thermodynamic stabilization following the same affinity order obtained in the docking simulations, without covalent-bond formation between both interacting beta-blockers in the best-ranked site. Lastly, experimental ultrasound density and velocity measurements were performed and allowed us to validate and corroborate the computational obtained results.
在这项工作中,评估了两种广泛使用的β受体阻滞剂药物(即醋丁洛尔和普萘洛尔)与最丰富的血浆纤维蛋白原蛋白之间最常见的多药理学药物相互作用事件之一。为此,使用分子对接和密度泛函理论(DFT)计算作为补充工具。对三个排名最高的结合部位的纤维蛋白原晶体学验证显示,构象有利的残基比例为 100%,完全没有受限的灵活性。在这三个部位中,基于结合部位可成药性和配体转运分析的自由能轨迹的结果都指出了最适合药物相互作用的生物物理环境部位。此外,药物-药物复合物的总稳定性主要受到空间能贡献的影响,这主要是基于与最佳排名部位中的关键残基(THR22:P 和 SER50:Q)的多个疏水接触。此外,DFT 计算表明,β受体阻滞剂药物-药物复合物具有自发的热力学稳定性,遵循与对接模拟中获得的相同亲和力顺序,而在最佳排名部位中,两个相互作用的β受体阻滞剂之间没有形成共价键。最后,进行了实验超声密度和速度测量,使我们能够验证和证实计算得到的结果。