College of Medicine, University of Sharjah, Sharjah, United Arab Emirates.
Sharjah Institute for Medical Research, University of Sharjah, Sharjah, United Arab Emirates.
PLoS One. 2020 Nov 30;15(11):e0242695. doi: 10.1371/journal.pone.0242695. eCollection 2020.
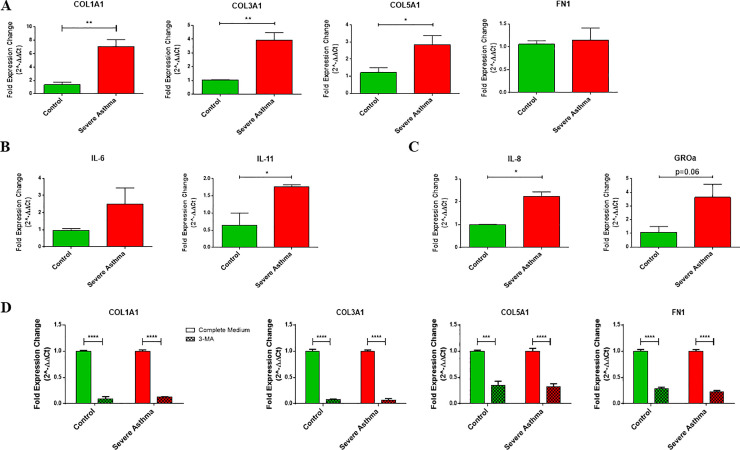
Sub-epithelial fibrosis is a characteristic feature of airway remodeling in asthma which correlates with disease severity. Current asthma medications are ineffective in treating fibrosis. In this study, we aimed to investigate the mitochondrial phenotype in fibroblasts isolated from airway biopsies of non-asthmatic and severe asthmatic subjects by examining mitophagy as a mechanism contributing to fibroblast persistence and thereby, fibrosis in severe asthma.
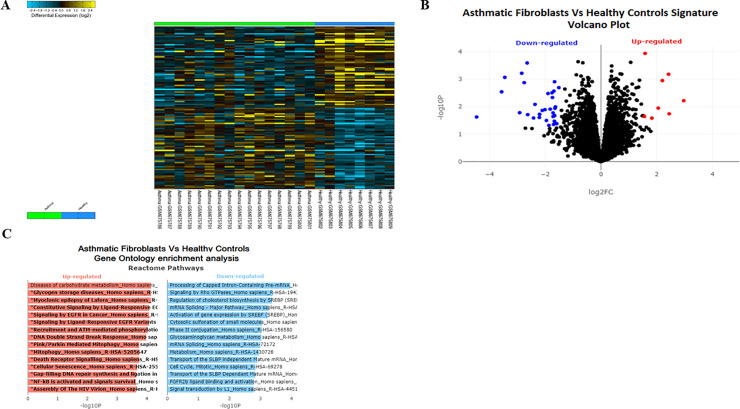
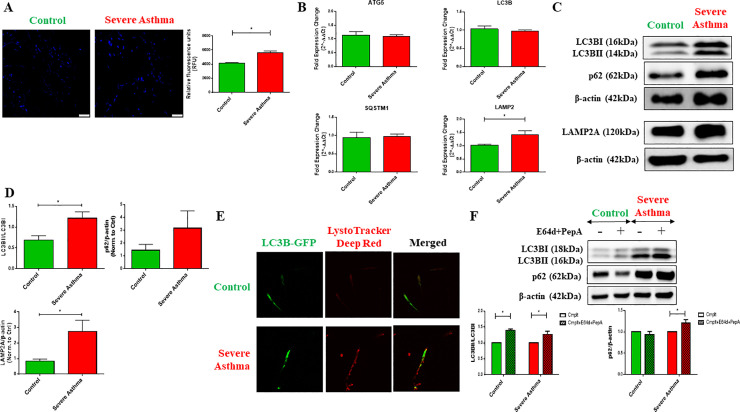
Bioinformatics analysis of publicly available transcriptomic data was performed to identify the top enriched pathways in asthmatic fibroblasts. Endogenous expression of mitophagy markers in severe asthmatic and non-asthmatic fibroblasts was determined using qRT-PCR, western blot and immunofluorescence. Mitophagy flux was examined by using lysosomal protease inhibitors, E64d and pepstatin A. Mitochondrial membrane potential and metabolic activity were also evaluated using JC-1 assay and MTT assay, respectively.
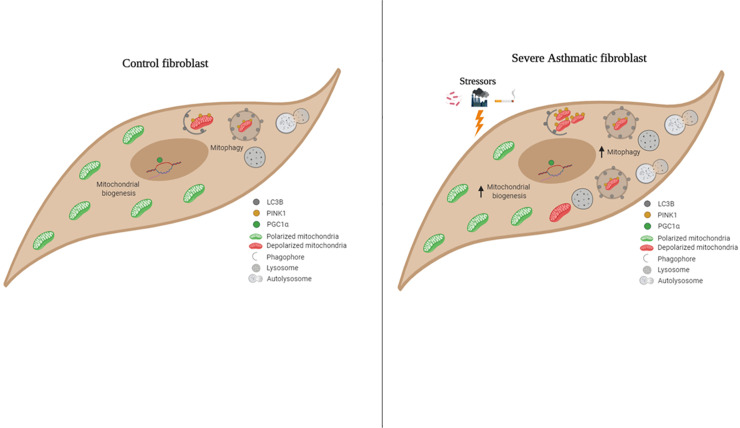
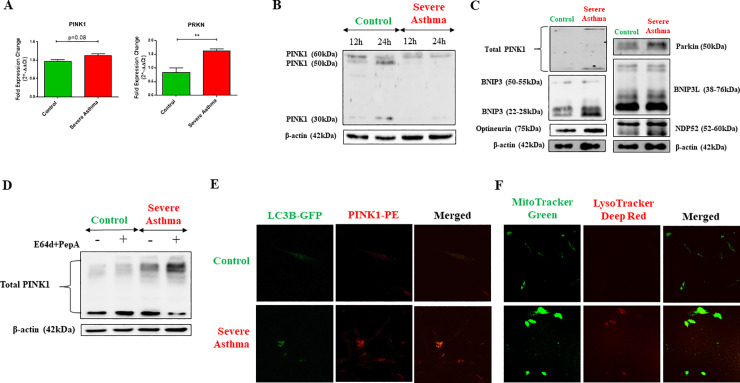
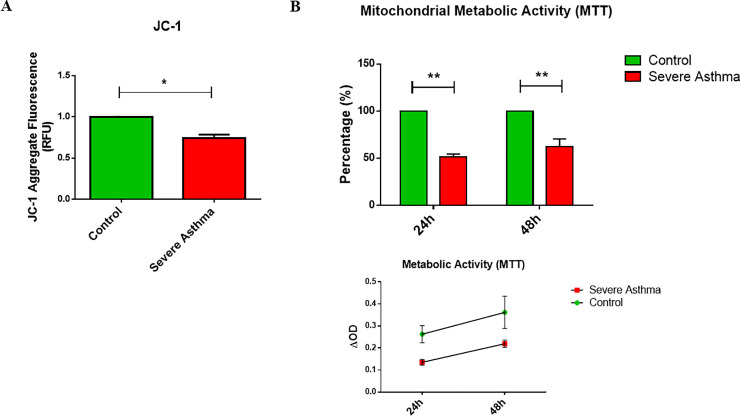
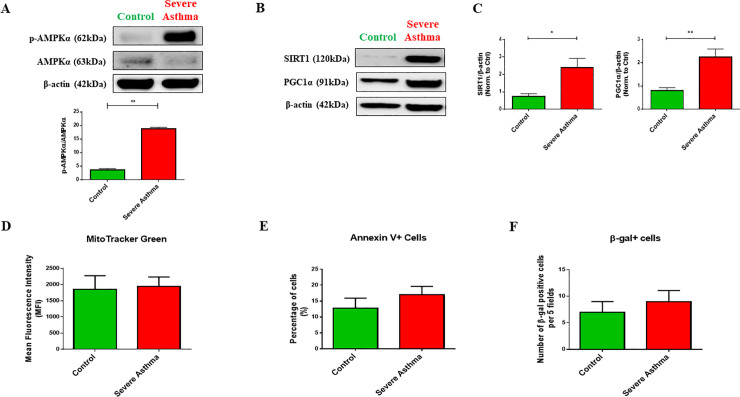
Bioinformatics analysis revealed the enrichment of Pink/Parkin-mediated mitophagy in asthmatic fibroblasts compared to healthy controls. In severe asthmatic fibroblasts, the differential expression of mitophagy genes, PINK1 and PRKN, was accompanied by the accumulation of PINK1, Parkin and other mitophagy proteins at baseline. The further accumulation of endogenous LC3BII, p62 and PINK1 in the presence of E64d and pepstatin A in severe asthmatic fibroblasts reinforced their enhanced mitophagy flux. Significantly reduced mitochondrial membrane potential and metabolic activity were also demonstrated at baseline confirming the impairment in mitochondrial function in severe asthmatic fibroblasts. Interestingly, these fibroblasts displayed neither an apoptotic nor senescent phenotype but a pro-fibrotic phenotype with an adaptive survival mechanism triggered by increased AMPKα phosphorylation and mitochondrial biogenesis.
Our results demonstrated a role for mitophagy in the pathogenesis of severe asthma where the enhanced turnover of damaged mitochondria may contribute to fibrosis in severe asthma by promoting the persistence and pro-fibrotic phenotype of fibroblasts.
气道重塑是哮喘的一个特征,其与疾病严重程度相关。目前的哮喘药物对纤维化无效。本研究旨在通过研究自噬作为导致成纤维细胞持续存在进而导致严重哮喘纤维化的机制,来研究非哮喘和严重哮喘患者气道活检中分离的成纤维细胞的线粒体表型。
对公开的转录组数据进行生物信息学分析,以确定哮喘成纤维细胞中富集的途径。使用 qRT-PCR、western blot 和免疫荧光法测定严重哮喘和非哮喘成纤维细胞中自噬标志物的内源性表达。通过使用溶酶体蛋白酶抑制剂 E64d 和胃蛋白酶抑制剂 A 检测自噬流。还使用 JC-1 测定法和 MTT 测定法分别评估线粒体膜电位和代谢活性。
生物信息学分析显示,与健康对照组相比,Pink/Parkin 介导的自噬在哮喘成纤维细胞中富集。在严重哮喘成纤维细胞中,自噬基因 PINK1 和 PRKN 的差异表达伴随着 PINK1、Parkin 和其他自噬蛋白在基线时的积累。在存在 E64d 和胃蛋白酶抑制剂 A 的情况下,严重哮喘成纤维细胞中内源性 LC3BII、p62 和 PINK1 的进一步积累增强了其增强的自噬通量。基线时还证实了严重哮喘成纤维细胞中线粒体功能受损,表现为线粒体膜电位和代谢活性显著降低。有趣的是,这些成纤维细胞既没有表现出凋亡也没有表现出衰老表型,而是表现出促纤维化表型,其具有由 AMPKα 磷酸化和线粒体生物发生触发的适应性生存机制。
我们的研究结果表明,自噬在严重哮喘的发病机制中起作用,受损线粒体的增强周转率可能通过促进成纤维细胞的持续存在和促纤维化表型导致严重哮喘的纤维化。