Friedrich-Baur-Institute, Department of Neurology, Ludwig-Maximilians-University, Ziemssenstrasse 1a, 80336, Munich, Germany.
Genetikum, Neu-Ulm, Germany.
J Neurol. 2021 May;268(5):1708-1720. doi: 10.1007/s00415-020-10328-1. Epub 2020 Dec 2.
Non-dystrophic myotonias (NDM) are heterogeneous diseases caused by mutations in CLCN1 and SCN4A. The study aimed to describe the clinical and genetic spectrum of NDM in a large German cohort.
We retrospectively identified all patients with genetically confirmed NDM diagnosed in our center. The following data were analyzed: demographics, family history, muscular features, cardiac involvement, CK, EMG, genotype, other tested genes, treatment perceived efficacy.
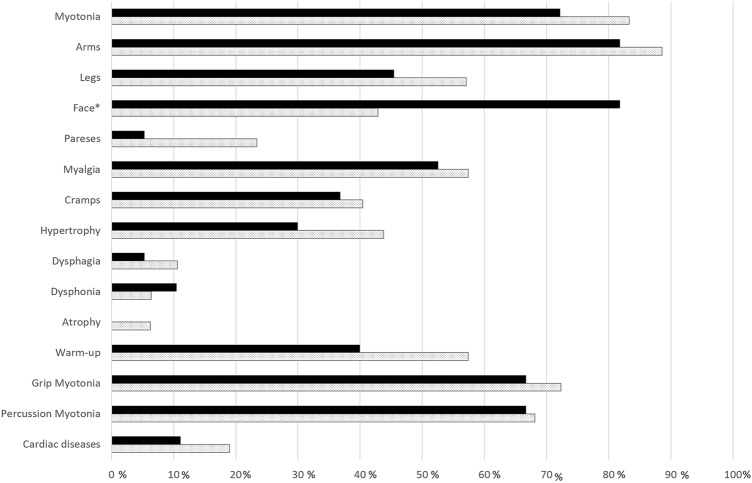
70 patients (age 40.2 years ± 14.9; 52.8% males) were included in our study (48 NDM-CLCN1, 22 NDM-SCN4A). The most frequent presenting symptoms were myotonia (NDM-CLCN1 83.3%, NDM-SCN4A 72.2%) and myalgia (NDM-CLCN1 57.4%, NDM-SCN4A 52.6%). Besides a more prominent facial involvement in NDM-SCN4A and cold-sensitivity in NDM-CLCN1, no other significant differences were observed between groups. Cardiac arrhythmia or conduction defects were documented in sixNDM-CLCN1 patients (three of them requiring a pacemaker) and one patient with NDM-SCN4A. CK was normal in 40% of patients. Myotonic runs in EMG were detected in 89.1% of CLCN1 and 78.9% of SCN4A. 50% of NDM-CLCN1 patients had the classic c.2680C>T (p.Arg894*) mutation. 12 new genetic variants are reported. About 50% of patients were not taking any anti-myotonic drug at the last follow-up. The anti-myotonic drugs with the best patient's perceived efficacy were mexiletine and lamotrigine.
This study highlights the relevant clinical overlap between NDM-CLCN1 and NDM-SCN4A patients and warrants the use of early and broad genetic investigation for the precise identification of the NDM subtype. Besides the clinical and genetic heterogeneity, the limited response to current anti-myotonic drugs constitutes a continuing challenge.
非营养不良性肌强直症(NDM)是由 CLCN1 和 SCN4A 基因突变引起的异质性疾病。本研究旨在描述一个大型德国队列中 NDM 的临床和遗传谱。
我们回顾性地确定了在我们中心诊断为基因证实的 NDM 的所有患者。分析了以下数据:人口统计学、家族史、肌肉特征、心脏受累、CK、EMG、基因型、其他测试基因、治疗效果感知。
共纳入 70 例患者(年龄 40.2 ± 14.9 岁;男性占 52.8%)(48 例 NDM-CLCN1,22 例 NDM-SCN4A)。最常见的首发症状为肌强直(NDM-CLCN1 83.3%,NDM-SCN4A 72.2%)和肌痛(NDM-CLCN1 57.4%,NDM-SCN4A 52.6%)。除了 NDM-SCN4A 患者更明显的面部受累和 NDM-CLCN1 患者的冷敏感性外,两组之间没有观察到其他显著差异。6 例 NDM-CLCN1 患者(其中 3 例需要起搏器)和 1 例 NDM-SCN4A 患者记录到心律失常或传导缺陷。40%的患者 CK 正常。在 89.1%的 CLCN1 和 78.9%的 SCN4A 患者的 EMG 中检测到肌强直发作。50%的 NDM-CLCN1 患者存在经典的 c.2680C>T(p.Arg894*)突变。报告了 12 种新的遗传变异。大约 50%的患者在最后一次随访时没有服用任何抗肌强直药物。患者感知效果最好的抗肌强直药物是美西律和拉莫三嗪。
本研究强调了 NDM-CLCN1 和 NDM-SCN4A 患者之间的相关临床重叠,并证明需要早期和广泛的遗传研究来精确确定 NDM 亚型。除了临床和遗传异质性外,目前抗肌强直药物的疗效有限仍然是一个持续的挑战。