Department of Medicine, Division of Endocrinology, David Geffen School of Medicine at UCLA, 650 Charles E. Young Dr., Los Angeles, CA, 90095, USA; Department of Molecular and Medical Pharmacology, David Geffen School of Medicine at UCLA, 650 Charles E. Young Dr., Los Angeles, CA, 90095, USA; Molecular Biology Institute at UCLA, Los Angeles, CA, 90095, USA.
Department of Medicine, Division of Endocrinology, David Geffen School of Medicine at UCLA, 650 Charles E. Young Dr., Los Angeles, CA, 90095, USA; Department of Chemistry and Biochemistry, UCLA, Los Angeles, CA, 90024, USA.
Mol Metab. 2021 Aug;50:101134. doi: 10.1016/j.molmet.2020.101134. Epub 2020 Dec 1.
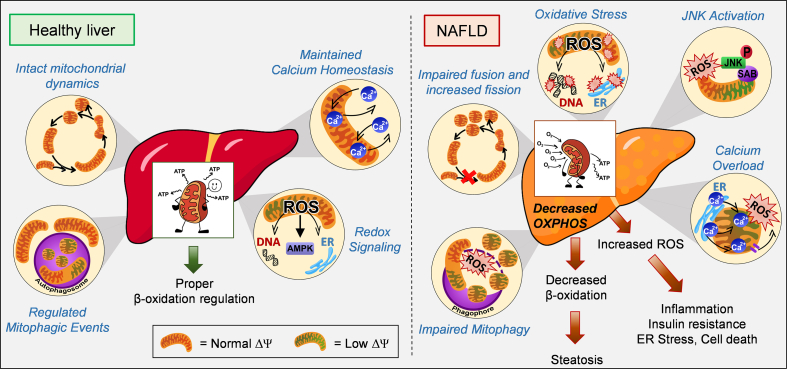
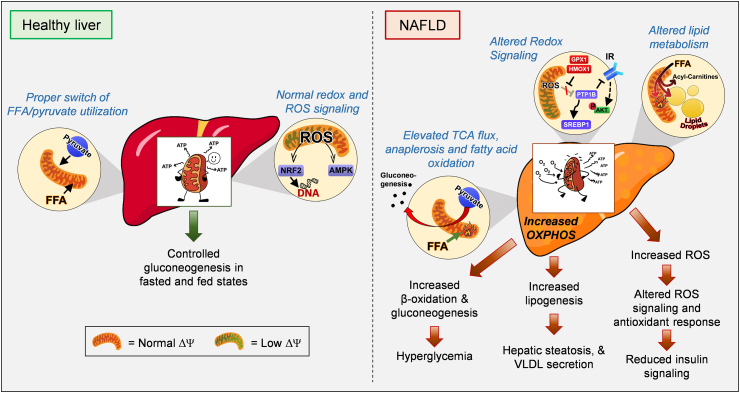
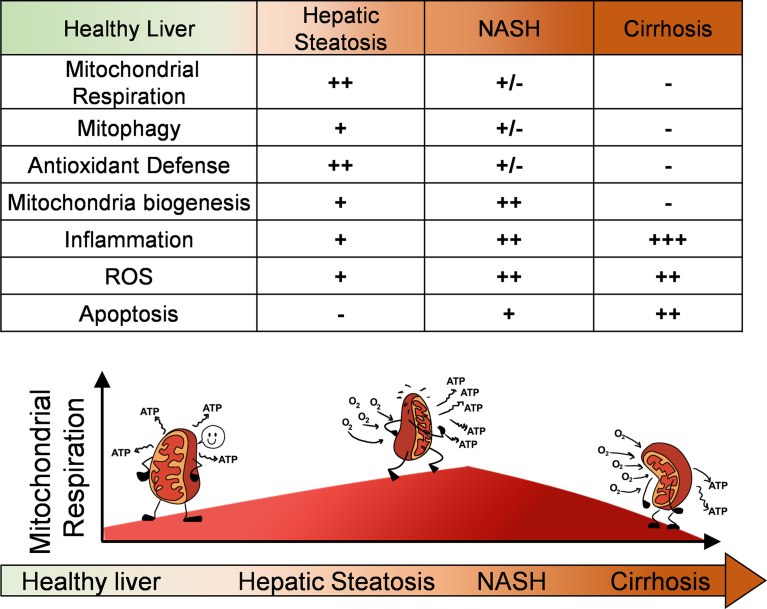
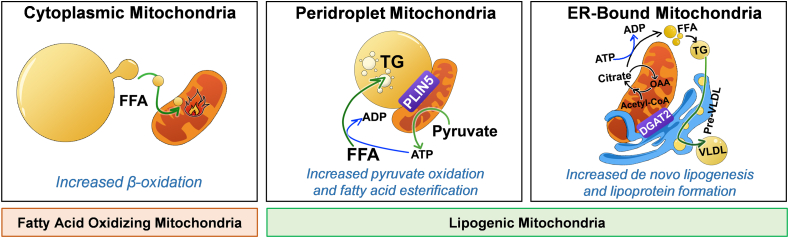
Mitochondrial oxidative function plays a key role in the development of non-alcoholic fatty liver disease (NAFLD) and insulin resistance (IR). Recent studies reported that fatty liver might not be a result of decreased mitochondrial fat oxidation caused by mitochondrial damage. Rather, NAFLD and IR induce an elevation in mitochondrial function that covers the increased demand for carbon intermediates and ATP caused by elevated lipogenesis and gluconeogenesis. Furthermore, mitochondria play a role in regulating hepatic insulin sensitivity and lipogenesis by modulating redox-sensitive signaling pathways.
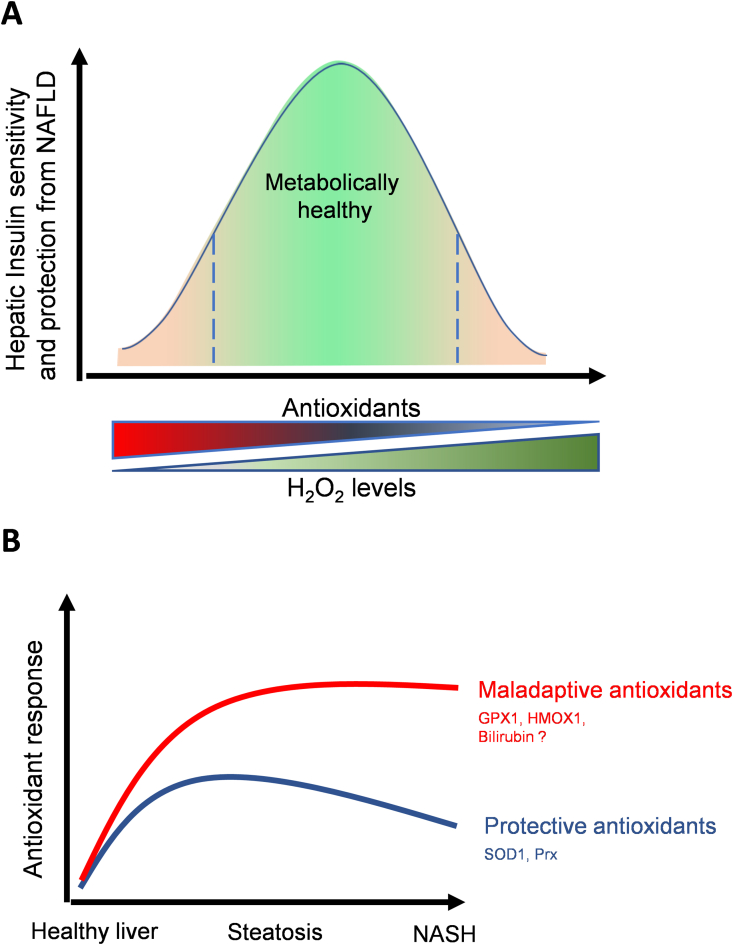
We review the contradictory studies indicating that NAFLD and hyperglycemia can either increase or decrease mitochondrial oxidative capacity in the liver. We summarize mechanisms regulating mitochondrial heterogeneity inside the same cell and discuss how these mechanisms may determine the role of mitochondria in NAFLD. We further discuss the role of endogenous antioxidants in controlling mitochondrial HO release and redox-mediated signaling. We describe the emerging concept that the subcellular location of cellular antioxidants is a key determinant of their effects on NAFLD.
The balance of fat oxidation versus accumulation depends on mitochondrial fuel preference rather than ATP-synthesizing respiration. As such, therapies targeting fuel preference might be more suitable for treating NAFLD. Similarly, suppressing maladaptive antioxidants, rather than interfering with physiological mitochondrial HO-mediated signaling, may allow the maintenance of intact hepatic insulin signaling in NAFLD. Exploration of the subcellular compartmentalization of different antioxidant systems and the unique functions of specific mitochondrial subpopulations may offer new intervention points to treat NAFLD.
线粒体氧化功能在非酒精性脂肪性肝病(NAFLD)和胰岛素抵抗(IR)的发展中起着关键作用。最近的研究报告称,脂肪肝可能不是由于线粒体损伤导致的线粒体脂肪氧化减少引起的。相反,NAFLD 和 IR 会引起线粒体功能升高,以满足脂肪生成和糖异生增加所导致的碳中间产物和 ATP 的增加需求。此外,线粒体通过调节氧化还原敏感信号通路在调节肝胰岛素敏感性和脂肪生成中发挥作用。
我们综述了相互矛盾的研究,这些研究表明,NAFLD 和高血糖症既可以增加也可以降低肝脏中线粒体的氧化能力。我们总结了调节同一细胞内线粒体异质性的机制,并讨论了这些机制如何决定线粒体在 NAFLD 中的作用。我们进一步讨论了内源性抗氧化剂在控制线粒体 HO 释放和氧化还原介导的信号中的作用。我们描述了一个新兴概念,即细胞抗氧化剂的亚细胞位置是决定其对 NAFLD 影响的关键决定因素。
脂肪氧化与积累的平衡取决于线粒体对燃料的偏好,而不是 ATP 合成呼吸。因此,针对燃料偏好的治疗方法可能更适合治疗 NAFLD。同样,抑制适应性不良的抗氧化剂,而不是干扰生理线粒体 HO 介导的信号,可能允许在 NAFLD 中维持完整的肝胰岛素信号。探索不同抗氧化系统的亚细胞区室化和特定线粒体亚群的独特功能可能为治疗 NAFLD 提供新的干预点。