Morais Sara, Oliveira Jorge, Lau Catarina, Pereira Mónica, Gonçalves Marta, Monteiro Catarina, Gonçalves Ana Rita, Matos Rui, Sampaio Marco, Cruz Eugénia, Freitas Inês, Santos Rosário, Lima Margarida
Setor de Trombose e Hemostase, Serviço de Hematologia Clínica, Hospital de Santo António (HSA), Centro Hospitalar Universitário do Porto (CHUP), Porto, Portugal.
Unidade Multidisciplinar de Investigação Biomédica, Instituto de Ciências Biomédicas, Universidade do Porto (UMIB/ICBAS/UP), Porto, Portugal.
PLoS One. 2020 Dec 4;15(12):e0235136. doi: 10.1371/journal.pone.0235136. eCollection 2020.
Rare pathogenic variants in either the ITGA2B or ITGB3 genes have been linked to autosomal dominant macrothrombocytopenia associated with abnormal platelet production and function, deserving the designation of Glanzmann Thrombasthenia-Like Syndrome (GTLS) or ITGA2B/ITGB3-related thrombocytopenia.
To describe a series of patients with familial macrothrombocytopenia and decreased expression of αIIbβ3 integrin due to defects in the ITGA2B or ITGB3 genes.
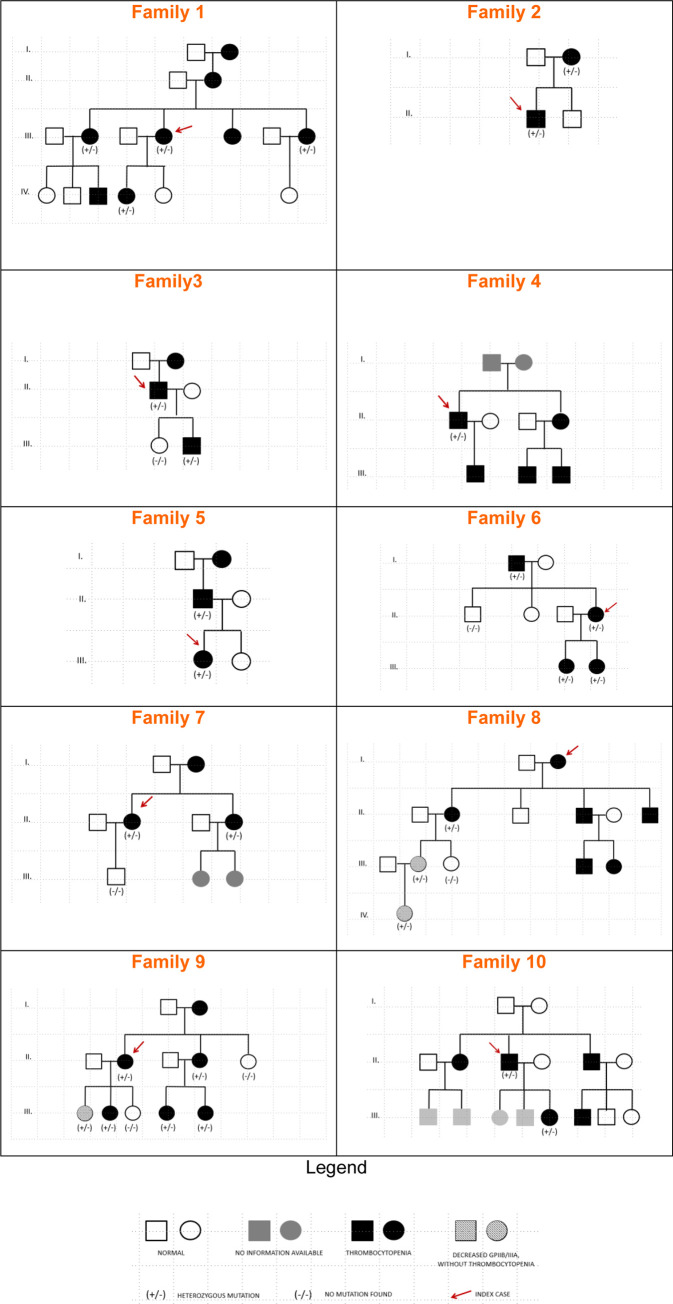
We reviewed the clinical and laboratory records of 10 Portuguese families with GTLS (33 patients and 11 unaffected relatives), including the functional and genetic defects.
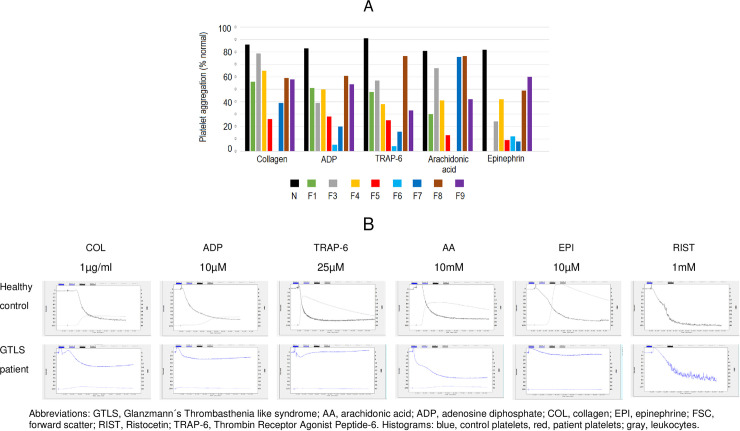
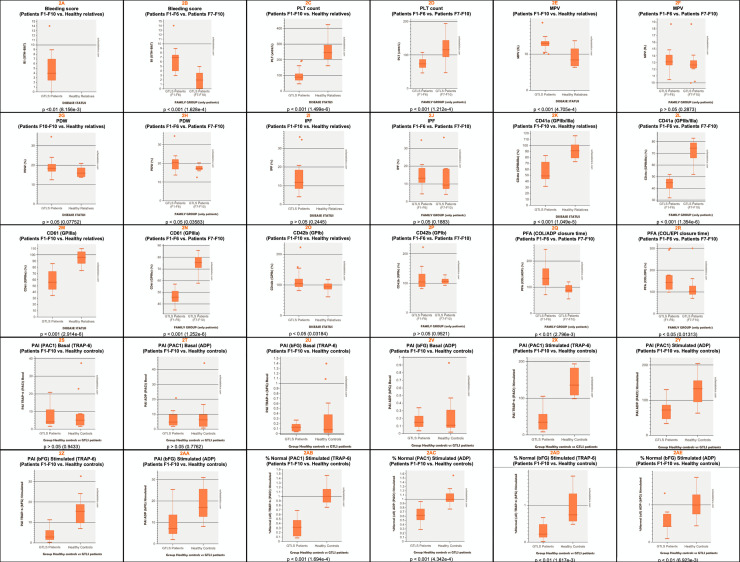
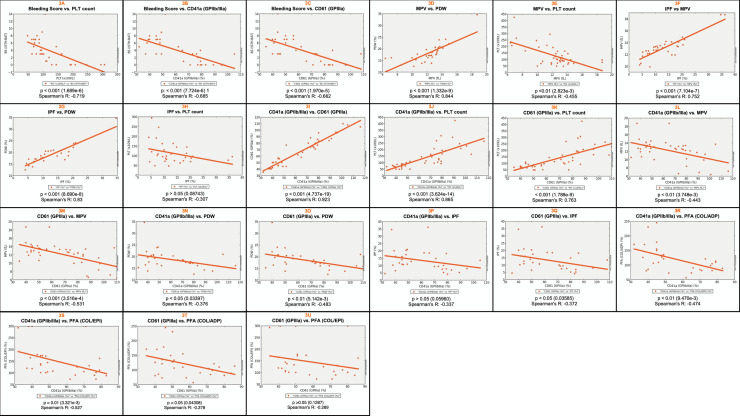
Patients had absent to moderate bleeding, macrothrombocytopenia, low αIIbβ3 expression, impaired platelet aggregation/ATP release to physiological agonists and low expression of activation-induced binding sites on αIIbβ3 (PAC-1) and receptor-induced binding sites on its ligand (bound fibrinogen), upon stimulation with TRAP-6 and ADP. Evidence for constitutive αIIbβ3 activation, occurred in 2 out of 9 patients from 8 families studied, but also in 2 out of 12 healthy controls. We identified 7 missense variants: 3 in ITGA2B (5 families), and 4 in ITGB3 (5 families). Three variants (αIIb: p.Arg1026Trp and p.Arg1026Gln and β3: p.Asp749His) were previously reported. The remaining (αIIb: p.Gly1007Val and β3: p.Thr746Pro, p.His748Pro and p.Arg760Cys) are new, expanding the αIIbβ3 defects associated with GTLS. The integration of the clinical and laboratory data allowed the identification of two GTLS subgroups, with distinct disease severity.
Previously reported ITGA2B and ITGB3 variants related to thrombocytopenia were clustered in a confined region of the membrane-proximal cytoplasmic domains, the inner membrane clasp. For the first time, variants are reported at the outer membrane clasp, at the transmembrane domain of αIIb, and at the membrane distal cytoplasmic domains of β3. This is the largest single-center series of inherited macrothrombocytopenia associated with αIIbβ3 variants published to date.
ITGA2B或ITGB3基因中的罕见致病变异与常染色体显性遗传性大血小板减少症相关,该病症伴有血小板生成和功能异常,值得被命名为类Glanzmann血小板无力症综合征(GTLS)或ITGA2B/ITGB3相关血小板减少症。
描述一系列因ITGA2B或ITGB3基因缺陷导致家族性大血小板减少症及αIIbβ3整合素表达降低的患者。
我们回顾了10个葡萄牙GTLS家族(33例患者和11名未患病亲属)的临床和实验室记录,包括功能和基因缺陷情况。
患者存在轻度至中度出血、大血小板减少症、αIIbβ3表达降低、血小板对生理性激动剂的聚集/ATP释放受损,以及在用TRAP-6和ADP刺激后,αIIbβ3上的激活诱导结合位点(PAC-1)和其配体上的受体诱导结合位点(结合纤维蛋白原)表达降低。在研究的8个家族的9例患者中有2例出现组成型αIIbβ3激活的证据,但在12名健康对照者中也有2例出现。我们鉴定出7个错义变异:3个在ITGA2B基因中(5个家族),4个在ITGB3基因中(5个家族)。其中3个变异(αIIb:p.Arg1026Trp和p.Arg1026Gln以及β3:p.Asp749His)先前已有报道。其余变异(αIIb:p.Gly1007Val和β3:p.Thr746Pro、p.His748Pro和p.Arg760Cys)是新发现的,扩展了与GTLS相关的αIIbβ3缺陷。临床和实验室数据的整合使得能够识别出两个疾病严重程度不同的GTLS亚组。
先前报道的与血小板减少症相关的ITGA2B和ITGB3变异集中在膜近端细胞质结构域的一个受限区域,即内膜扣。首次报道了在外膜扣、αIIb的跨膜结构域以及β3的膜远端细胞质结构域出现的变异。这是迄今为止发表的与αIIbβ3变异相关的遗传性大血小板减少症的最大单中心系列研究。