Salazar-Silva Rodrigo, Dantas Vitor Lima Goes, Alves Leandro Ucela, Batissoco Ana Carla, Oiticica Jeanne, Lawrence Elizabeth A, Kawafi Abdelwahab, Yang Yushi, Nicastro Fernanda Stávale, Novaes Beatriz Caiuby, Hammond Chrissy, Kague Erika, Mingroni-Netto Regina Célia
Centro de Pesquisas sobre o Genoma Humano e Células-Tronco, Departamento de Genética e Biologia Evolutiva, Instituto de Biociências, Universidade de São Paulo, 05508-090, São Paulo, Brazil.
Laboratório de Otorrinolaringologia/LIM32 -Hospital das Clínicas, Faculdade de Medicina, Universidade de São Paulo , 01246-903, São Paulo, Brazil.
Hum Mol Genet. 2021 Jan 21;29(22):3691-3705. doi: 10.1093/hmg/ddaa240.
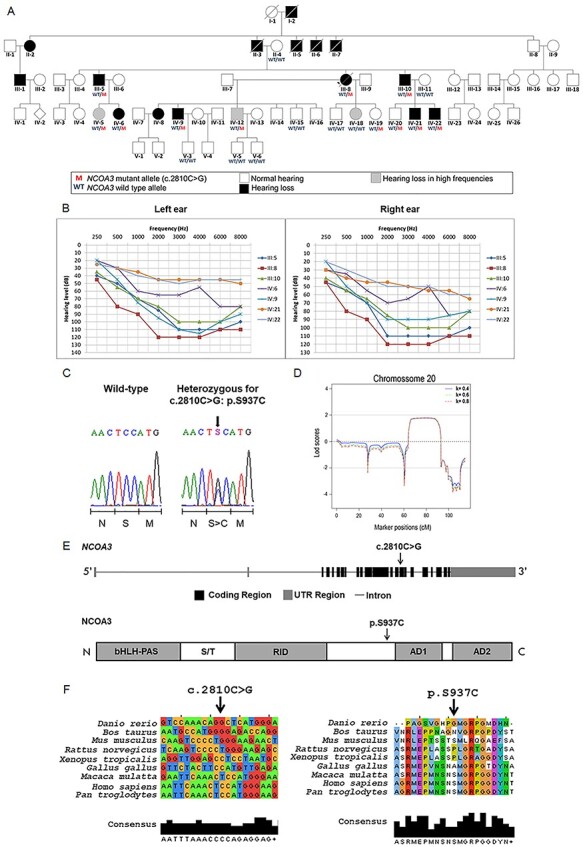
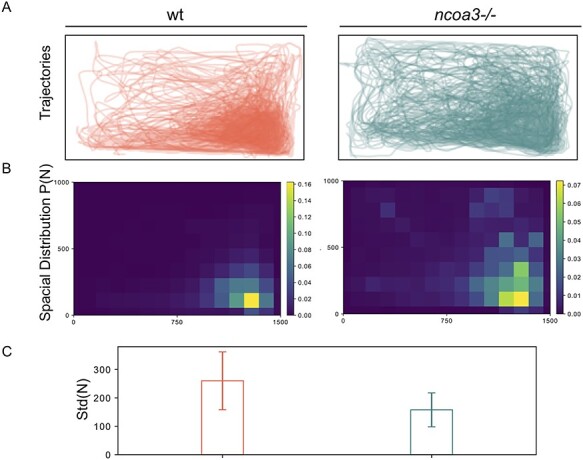
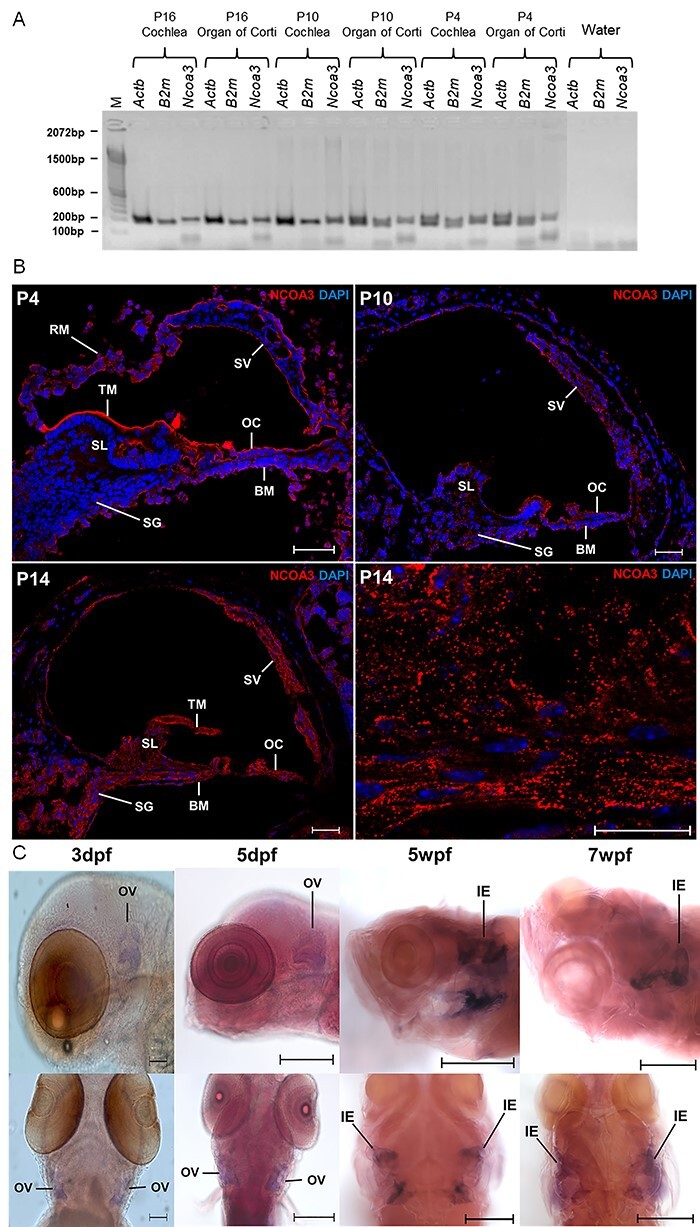
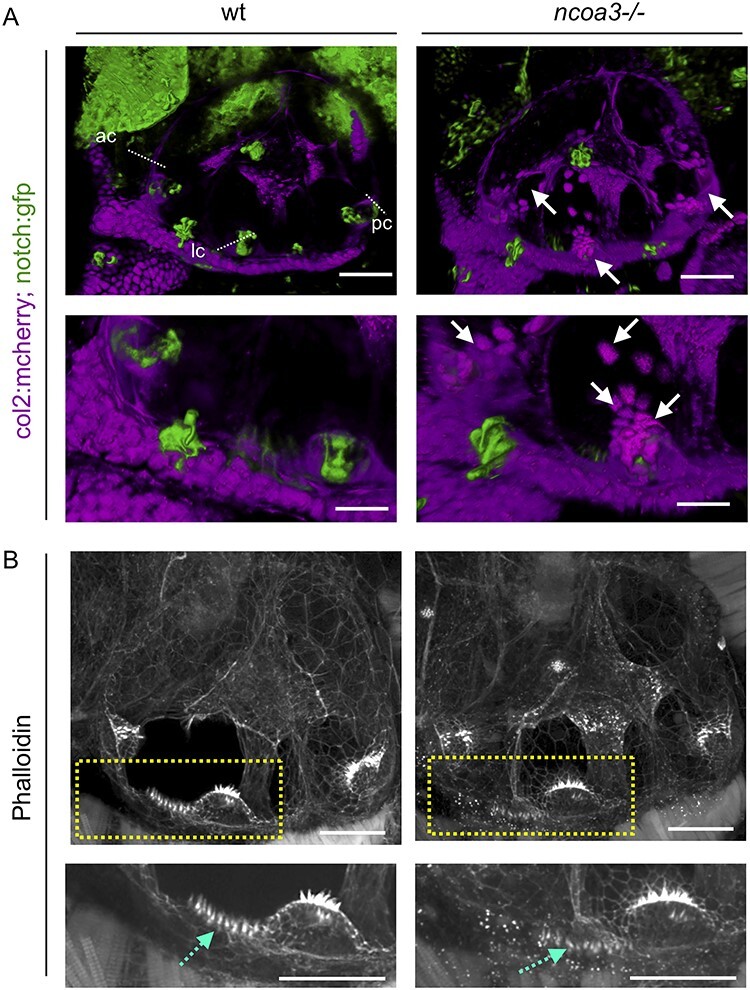
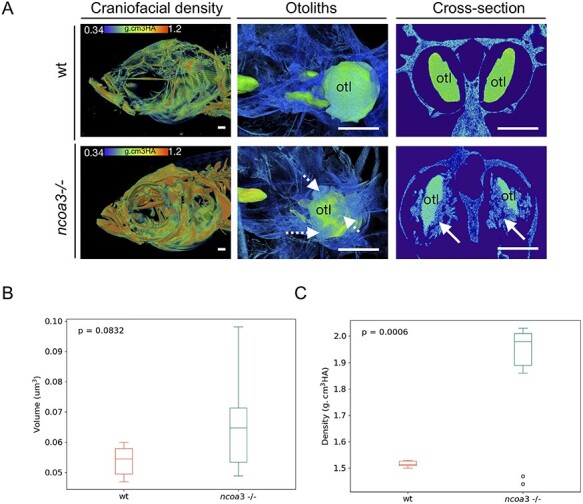
Hearing loss is a frequent sensory impairment in humans and genetic factors account for an elevated fraction of the cases. We have investigated a large family of five generations, with 15 reported individuals presenting non-syndromic, sensorineural, bilateral and progressive hearing loss, segregating as an autosomal dominant condition. Linkage analysis, using SNP-array and selected microsatellites, identified a region of near 13 cM in chromosome 20 as the best candidate to harbour the causative mutation. After exome sequencing and filtering of variants, only one predicted deleterious variant in the NCOA3 gene (NM_181659, c.2810C > G; p.Ser937Cys) fit in with our linkage data. RT-PCR, immunostaining and in situ hybridization showed expression of ncoa3 in the inner ear of mice and zebrafish. We generated a stable homozygous zebrafish mutant line using the CRISPR/Cas9 system. ncoa3-/- did not display any major morphological abnormalities in the ear, however, anterior macular hair cells showed altered orientation. Surprisingly, chondrocytes forming the ear cartilage showed abnormal behaviour in ncoa3-/-, detaching from their location, invading the ear canal and blocking the cristae. Adult mutants displayed accumulation of denser material wrapping the otoliths of ncoa3-/- and increased bone mineral density. Altered zebrafish swimming behaviour corroborates a potential role of ncoa3 in hearing loss. In conclusion, we identified a potential candidate gene to explain hereditary hearing loss, and our functional analyses suggest subtle and abnormal skeletal behaviour as mechanisms involved in the pathogenesis of progressive sensory function impairment.
听力损失是人类常见的感觉障碍,遗传因素在其中所占比例较高。我们研究了一个五代大家庭,其中15名成员患有非综合征性、感音神经性、双侧性和进行性听力损失,呈常染色体显性遗传。通过单核苷酸多态性阵列(SNP-array)和选定的微卫星进行连锁分析,确定20号染色体上一个近13厘摩的区域是最有可能携带致病突变的候选区域。在对变异进行外显子组测序和筛选后,只有NCOA3基因中的一个预测有害变异(NM_181659,c.2810C>G;p.Ser937Cys)与我们的连锁数据相符。逆转录聚合酶链反应(RT-PCR)、免疫染色和原位杂交显示ncoa3在小鼠和斑马鱼的内耳中表达。我们使用CRISPR/Cas9系统构建了一个稳定的纯合斑马鱼突变系。ncoa3-/-在耳朵中未表现出任何主要形态异常,然而,前黄斑毛细胞的方向发生了改变。令人惊讶的是,形成耳软骨的软骨细胞在ncoa3-/-中表现出异常行为,从其位置脱离,侵入耳道并阻塞嵴。成年突变体显示包裹ncoa3-/-耳石的致密物质积累,骨矿物质密度增加。斑马鱼游泳行为的改变证实了ncoa3在听力损失中的潜在作用。总之,我们确定了一个可能解释遗传性听力损失的候选基因,我们的功能分析表明,微妙和异常的骨骼行为是进行性感官功能障碍发病机制中的相关机制。