Pekgül Faruk, Eroğlu-Ertuğrul Nesibe Gevher, Bekircan-Kurt Can Ebru, Erdem-Ozdamar Sevim, Çetinkaya Arda, Tan Ersin, Konuşkan Bahadır, Karaağaoğlu Ergun, Topçu Meral, Akarsu Nurten Ayşe, Oguz Kader K, Anlar Banu, Özkara Hatice Asuman
Department of Medical Biochemistry, Faculty of Medicine, Hacettepe University, 06230 Ankara, Turkey.
Department of Pediatric Neurology, Faculty of Medicine, Hacettepe University, 06230 Ankara, Turkey.
Mol Genet Metab Rep. 2020 Dec 11;25:100688. doi: 10.1016/j.ymgmr.2020.100688. eCollection 2020 Dec.
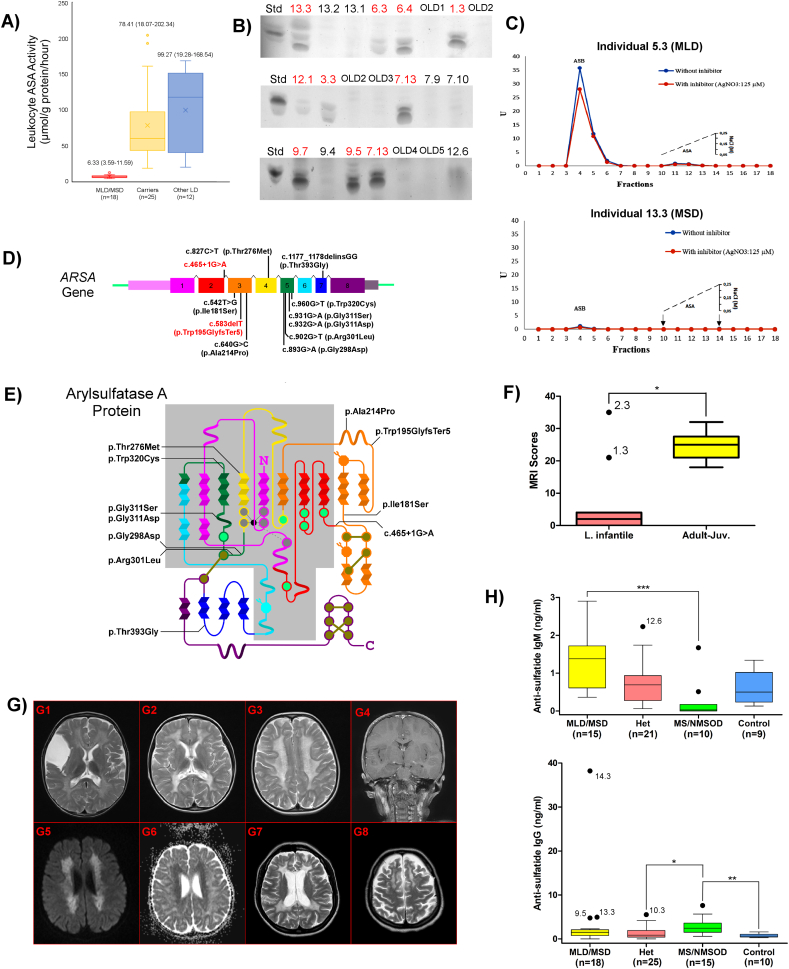
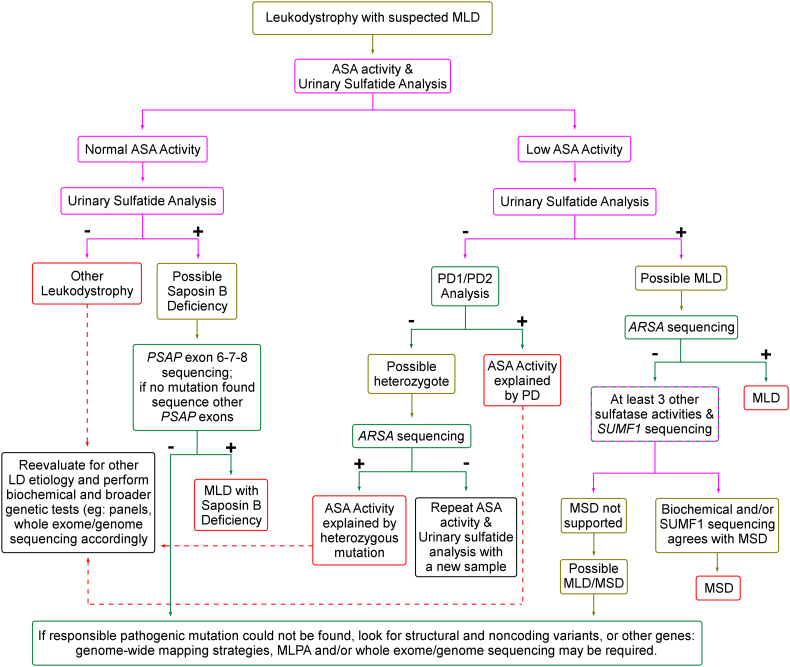
Metachromatic leukodystrophy (MLD) is a glycosphingolipid storage disease caused by deficiency of the lysosomal enzyme arylsulfatase A (ASA) or its activator protein saposin B. MLD can affect all age groups in severity varying from a severe fatal form to milder adult onset forms. Diagnosis is usually made by measuring leukocyte ASA activity. However, this test can give false negative or false positive laboratory results due to pseudodeficiency of ASA and saposin B deficiency, respectively. Therefore, we aimed to evaluate patients with suspected MLD in a Turkish population by comprehensive clinical, biochemical, radiological, and genetic analyses for molecular and phenotypic characterization. We analyzed 28 suspected MLD patients and 41 relatives from 24 families. ASA activity was found to be decreased in 21 of 28 patients. Sixteen patients were diagnosed as MLD (11 late infantile, 2 juvenile and 3 adult types), 2 MSD, 2 pseudodeficiency (PD) and the remaining 8 patients were diagnosed as having other leukodystrophies. Enzyme analysis showed that the age of onset of MLD did not correlate with residual ASA activity. Sequence analysis showed 11 mutations in , of which 4 were novel (p.Trp195GlyfsTer5, p.Gly298Asp, p.Arg301Leu, and p.Gly311Asp), and 2 mutations in causing multiple sulfatase deficiency, and confirmed the diagnosis of MLD in 2 presymptomatic relatives. All individuals with confirmed mutations had low ASA activity and urinary sulfatide excretion. Intra- and inter-familial variability was high for the same missense genotypes, indicating the contribution of other factors to disease expression. Imaging findings were evaluated through a modified brain MRI scoring system which indicated patients with protein-truncating mutations had more severe MRI findings and late-infantile disease onset. MRI findings were not specific for the diagnosis. Anti-sulfatide IgM was similar to control subjects, and IgG, elevated in multiple sulfatase deficiency. In conclusion, the knowledge on the biochemical, clinical and genetic basis of MLD was expanded, a modified diagnostic laboratory algorithm for MLD based on integrated evaluation of ASA activity, urinary sulfatide excretion and genetic tests was devised.
异染性脑白质营养不良(MLD)是一种鞘糖脂贮积病,由溶酶体酶芳基硫酸酯酶A(ASA)或其激活蛋白鞘脂激活蛋白B缺乏所致。MLD可影响所有年龄组,严重程度从严重致命型到较轻微的成人发病型不等。诊断通常通过检测白细胞ASA活性来进行。然而,由于分别存在ASA假缺陷和鞘脂激活蛋白B缺乏,该检测可能会给出假阴性或假阳性的实验室结果。因此,我们旨在通过全面的临床、生化、放射学和基因分析对土耳其人群中疑似MLD的患者进行分子和表型特征鉴定。我们分析了来自24个家庭的28例疑似MLD患者和41名亲属。发现28例患者中有21例ASA活性降低。16例患者被诊断为MLD(11例晚婴型、2例青少年型和3例成人型),2例为MSD,2例为假缺陷(PD),其余8例患者被诊断为患有其他脑白质营养不良。酶分析表明,MLD的发病年龄与残余ASA活性无关。序列分析显示, 中有11个突变,其中4个是新突变(p.Trp195GlyfsTer5、p.Gly298Asp、p.Arg301Leu和p.Gly311Asp), 中有2个突变导致多种硫酸酯酶缺乏,并在2名症状前亲属中确诊为MLD。所有确诊有突变的个体ASA活性均较低且尿硫脂排泄增加。对于相同的错义基因型,家族内和家族间的变异性都很高,这表明其他因素对疾病表达有影响。通过改良的脑MRI评分系统对影像学结果进行评估,该系统表明具有蛋白质截短突变的患者MRI表现更严重且发病为晚婴型。MRI表现对诊断不具有特异性。抗硫脂IgM与对照组相似,而IgG在多种硫酸酯酶缺乏时升高。总之,扩展了对MLD生化、临床和遗传基础的认识,设计了一种基于ASA活性、尿硫脂排泄和基因检测综合评估的改良MLD诊断实验室算法。