School of Life Sciences, Ulsan National Institute of Science and Technology, Ulsan, Republic of Korea.
Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea.
PLoS Biol. 2020 Dec 23;18(12):e3001002. doi: 10.1371/journal.pbio.3001002. eCollection 2020 Dec.
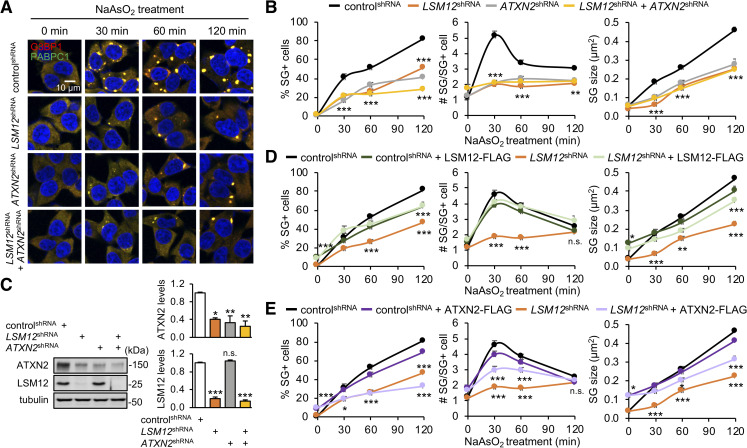
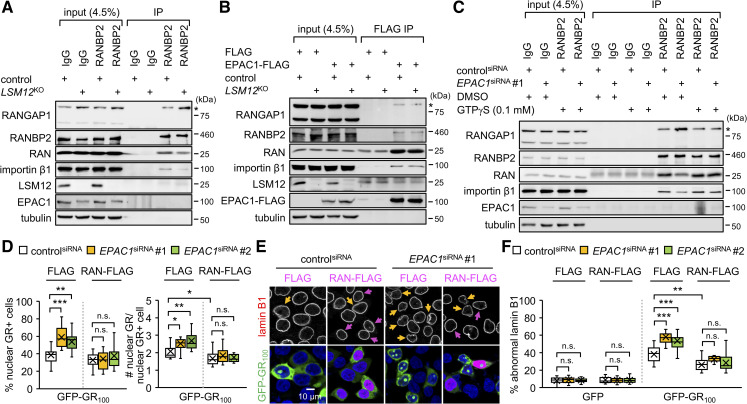
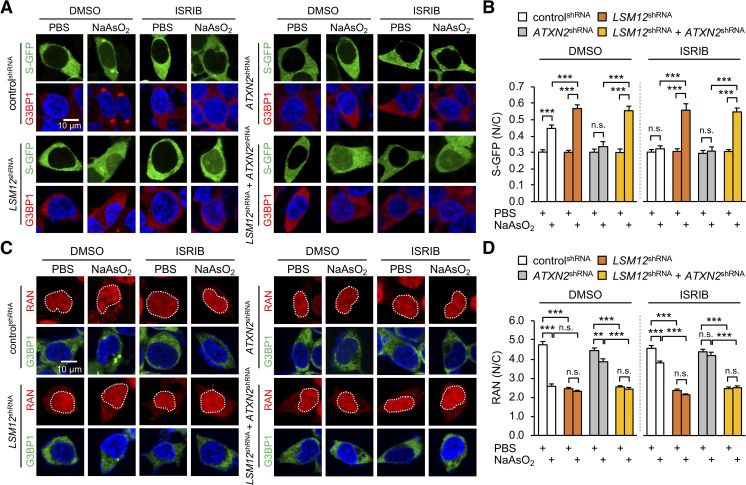
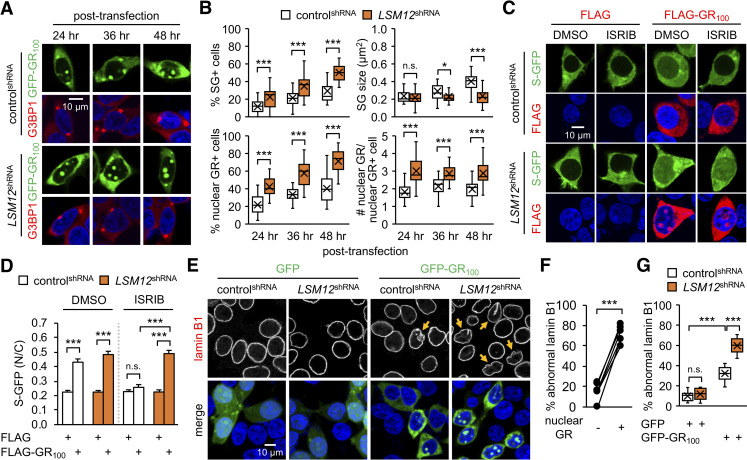
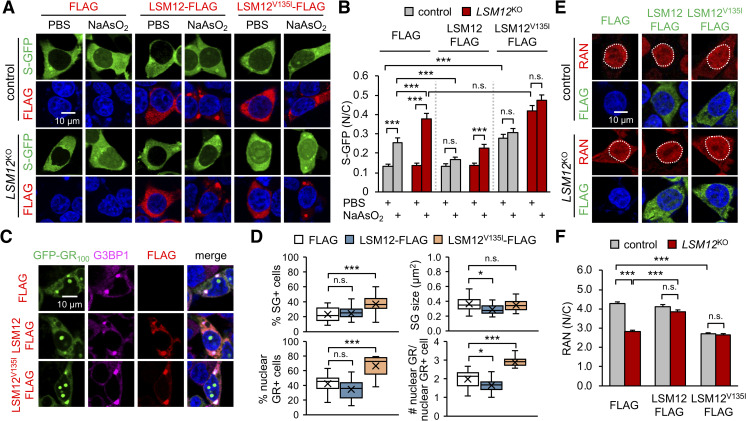
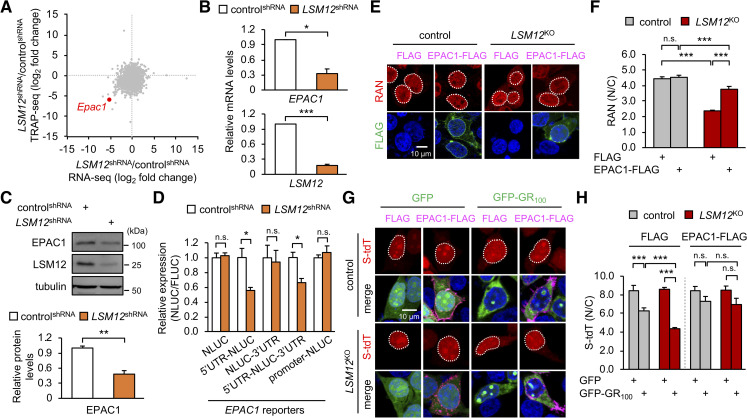
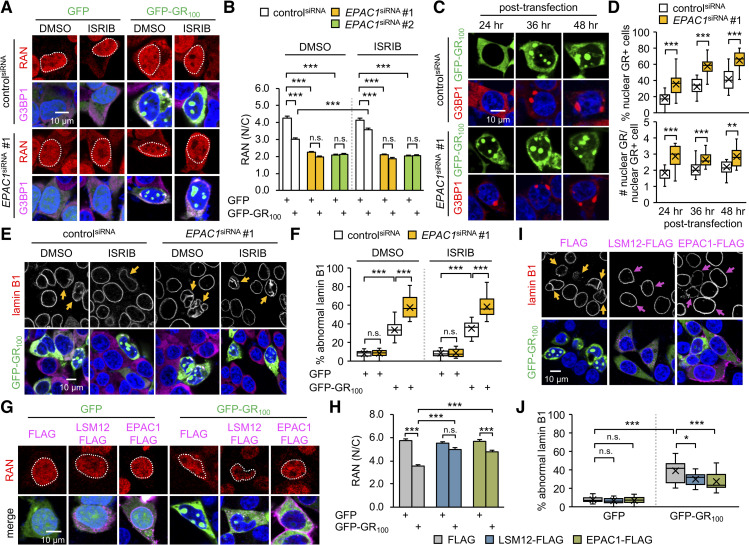
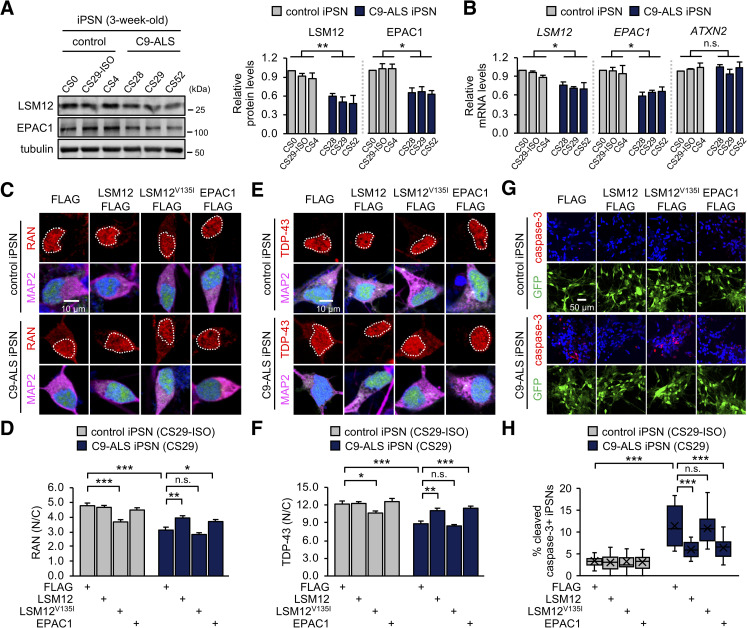
Nucleocytoplasmic transport (NCT) defects have been implicated in neurodegenerative diseases such as C9ORF72-associated amyotrophic lateral sclerosis and frontotemporal dementia (C9-ALS/FTD). Here, we identify a neuroprotective pathway of like-Sm protein 12 (LSM12) and exchange protein directly activated by cyclic AMP 1 (EPAC1) that sustains the nucleocytoplasmic RAN gradient and thereby suppresses NCT dysfunction by the C9ORF72-derived poly(glycine-arginine) protein. LSM12 depletion in human neuroblastoma cells aggravated poly(GR)-induced impairment of NCT and nuclear integrity while promoting the nuclear accumulation of poly(GR) granules. In fact, LSM12 posttranscriptionally up-regulated EPAC1 expression, whereas EPAC1 overexpression rescued the RAN gradient and NCT defects in LSM12-deleted cells. C9-ALS patient-derived neurons differentiated from induced pluripotent stem cells (C9-ALS iPSNs) displayed low expression of LSM12 and EPAC1. Lentiviral overexpression of LSM12 or EPAC1 indeed restored the RAN gradient, mitigated the pathogenic mislocalization of TDP-43, and suppressed caspase-3 activation for apoptosis in C9-ALS iPSNs. EPAC1 depletion biochemically dissociated RAN-importin β1 from the cytoplasmic nuclear pore complex, thereby dissipating the nucleocytoplasmic RAN gradient essential for NCT. These findings define the LSM12-EPAC1 pathway as an important suppressor of the NCT-related pathologies in C9-ALS/FTD.
核质转运 (NCT) 缺陷与神经退行性疾病有关,如 C9ORF72 相关肌萎缩侧索硬化症和额颞叶痴呆 (C9-ALS/FTD)。在这里,我们确定了一种像 Sm 蛋白 12 (LSM12) 和环 AMP 直接激活的交换蛋白 1 (EPAC1) 的神经保护途径,该途径维持核质 RAN 梯度,从而抑制由 C9ORF72 衍生的多 (甘氨酸-精氨酸) 蛋白引起的 NCT 功能障碍。人神经母细胞瘤细胞中 LSM12 的耗竭加剧了多 (GR) 诱导的 NCT 和核完整性损伤,同时促进了多 (GR) 颗粒的核积累。事实上,LSM12 转录后上调了 EPAC1 的表达,而 EPAC1 的过表达挽救了 LSM12 缺失细胞中的 RAN 梯度和 NCT 缺陷。来自诱导多能干细胞的 C9-ALS 患者源性神经元 (C9-ALS iPSNs) 显示 LSM12 和 EPAC1 的表达水平较低。慢病毒过表达 LSM12 或 EPAC1 确实恢复了 RAN 梯度,减轻了 TDP-43 的致病性定位错误,并抑制了 C9-ALS iPSNs 中的 caspase-3 激活以促进细胞凋亡。EPAC1 的耗竭从细胞质核孔复合物中生化分离 RAN-importin β1,从而破坏了 NCT 所必需的核质 RAN 梯度。这些发现将 LSM12-EPAC1 途径定义为 C9-ALS/FTD 中与 NCT 相关的病理学的重要抑制剂。