Milanetti Edoardo, Miotto Mattia, Di Rienzo Lorenzo, Monti Michele, Gosti Giorgio, Ruocco Giancarlo
Department of Physics, Sapienza University, Piazzale Aldo Moro 5, 00185 Rome, Italy.
Center for Life Nanoscience, Istituto Italiano di Tecnologia, Viale Regina Elena 291, 00161 Rome, Italy.
Comput Struct Biotechnol J. 2020 Dec 4;19:29-36. doi: 10.1016/j.csbj.2020.11.051. eCollection 2021.
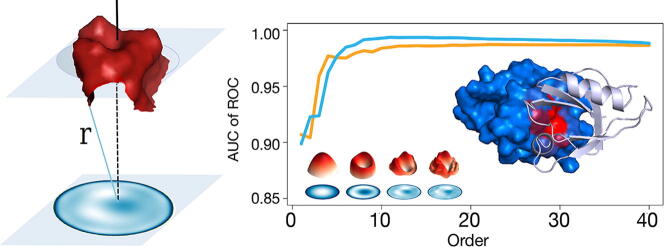
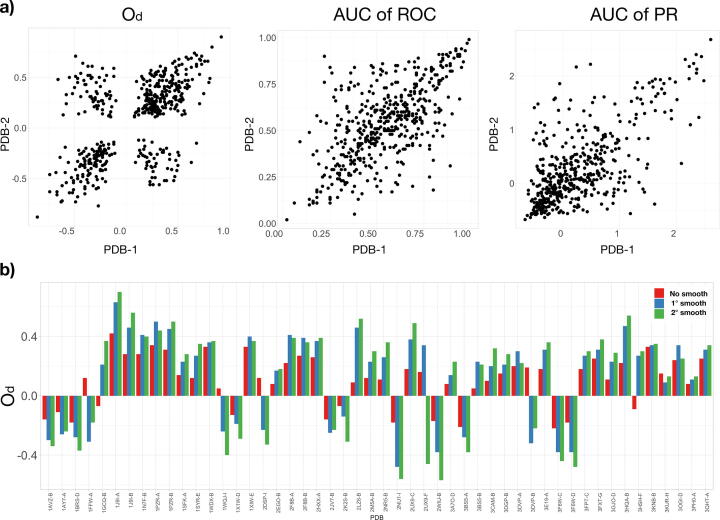
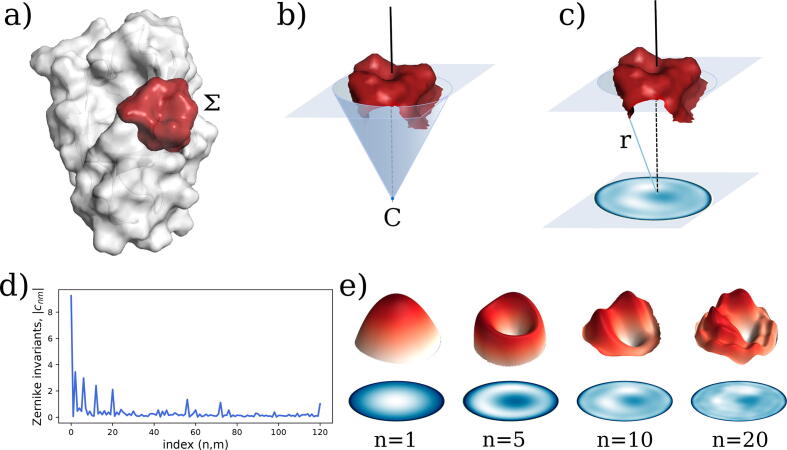
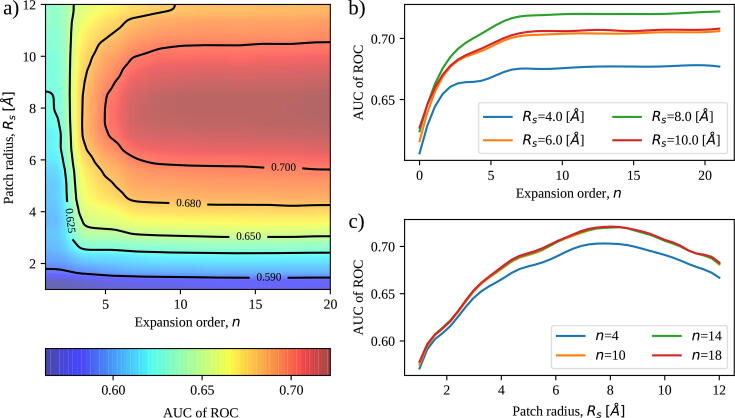
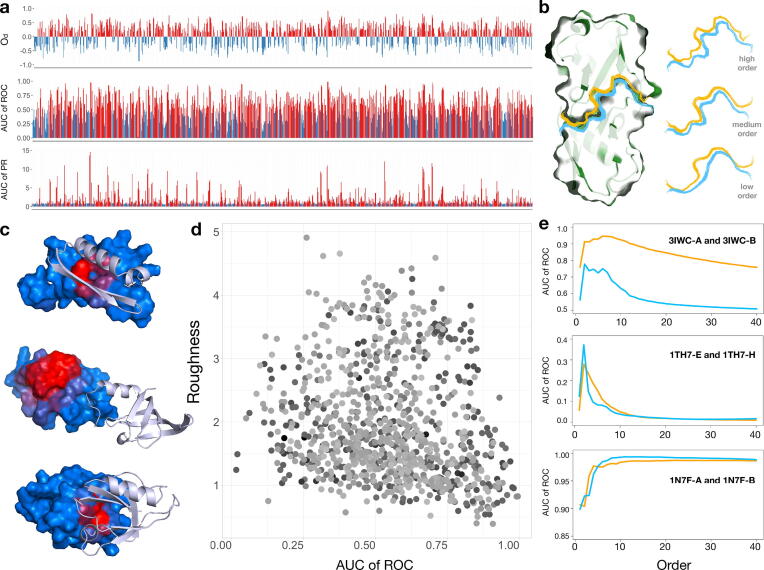
We present a method for efficiently and effectively assessing whether and where two proteins can interact with each other to form a complex. This is still largely an open problem, even for those relatively few cases where the 3D structure of both proteins is known. In fact, even if much of the information about the interaction is encoded in the chemical and geometric features of the structures, the set of possible contact patches and of their relative orientations are too large to be computationally affordable in a reasonable time, thus preventing the compilation of reliable interactome. Our method is able to rapidly and quantitatively measure the geometrical shape complementarity between interacting proteins, comparing their molecular iso-electron density surfaces expanding the surface patches in term of 2D Zernike polynomials. We first test the method against the real binding region of a large dataset of known protein complexes, reaching a success rate of 0.72. We then apply the method for the blind recognition of binding sites, identifying the real region of interaction in about of the analyzed cases. Finally, we investigate how the efficiency in finding the right binding region depends on the surface roughness as a function of the expansion order.
我们提出了一种方法,用于高效且有效地评估两种蛋白质是否以及在何处能够相互作用形成复合物。即使对于两种蛋白质的三维结构均已知的相对较少的情况而言,这在很大程度上仍是一个悬而未决的问题。实际上,即便关于相互作用的许多信息都编码在结构的化学和几何特征中,但可能的接触区域及其相对取向的集合过于庞大,以至于在合理时间内通过计算难以承受,从而阻碍了可靠相互作用组的编制。我们的方法能够快速且定量地测量相互作用蛋白质之间的几何形状互补性,通过二维泽尼克多项式扩展表面区域来比较它们的分子等电子密度表面。我们首先针对一大组已知蛋白质复合物的真实结合区域测试该方法,成功率达到0.72。然后我们将该方法应用于结合位点的盲目识别,在约[此处原文缺失具体比例]的分析案例中识别出真实的相互作用区域。最后,我们研究找到正确结合区域的效率如何取决于作为扩展阶数函数的表面粗糙度。