Università di Bologna, Bologna, Italy.
Center for Life Nano & Neuroscience, Italian Institute of Technology, Rome, Italy.
PLoS One. 2022 Apr 14;17(4):e0266004. doi: 10.1371/journal.pone.0266004. eCollection 2022.
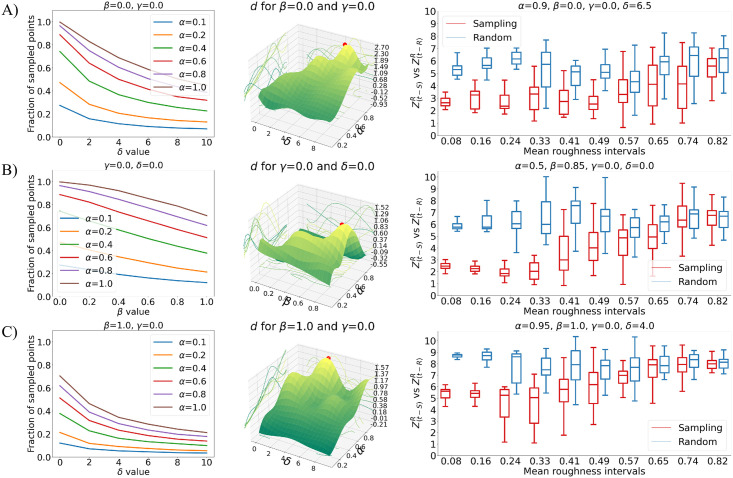
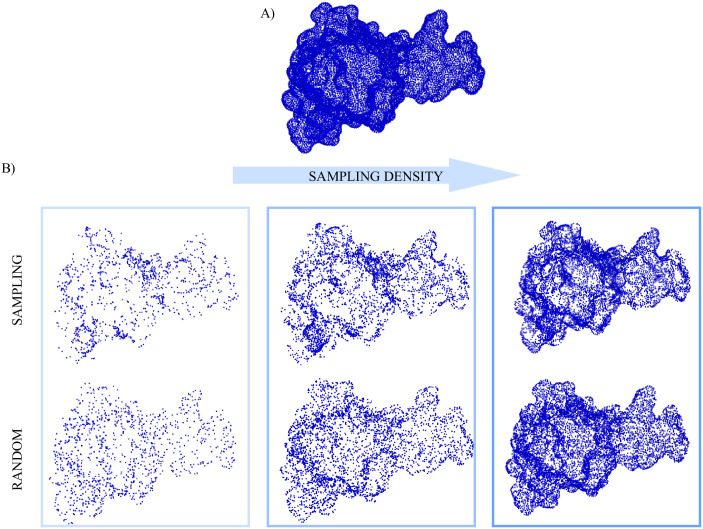
Most proteins perform their biological function by interacting with one or more molecular partners. In this respect, characterizing local features of the molecular surface, that can potentially be involved in the interaction with other molecules, represents a step forward in the investigation of the mechanisms of recognition and binding between molecules. Predictive methods often rely on extensive samplings of molecular patches with the aim to identify hot spots on the surface. In this framework, analysis of large proteins and/or many molecular dynamics frames is often unfeasible due to the high computational cost. Thus, finding optimal ways to reduce the number of points to be sampled maintaining the biological information (including the surface shape) carried by the molecular surface is pivotal. In this perspective, we here present a new theoretical and computational algorithm with the aim of defining a set of molecular surfaces composed of points not uniformly distributed in space, in such a way as to maximize the information of the overall shape of the molecule by minimizing the number of total points. We test our procedure's ability in recognizing hot-spots by describing the local shape properties of portions of molecular surfaces through a recently developed method based on the formalism of 2D Zernike polynomials. The results of this work show the ability of the proposed algorithm to preserve the key information of the molecular surface using a reduced number of points compared to the complete surface, where all points of the surface are used for the description. In fact, the methodology shows a significant gain of the information stored in the sampling procedure compared to uniform random sampling.
大多数蛋白质通过与一个或多个分子伴侣相互作用来发挥其生物功能。在这方面,描述分子表面局部特征,这些特征可能参与与其他分子的相互作用,是研究分子识别和结合机制的一个重要步骤。预测方法通常依赖于对分子斑块的广泛采样,目的是识别表面上的热点。在这个框架内,由于计算成本高,分析大型蛋白质和/或许多分子动力学帧通常是不可行的。因此,找到优化的方法来减少要采样的点数,同时保持分子表面所携带的生物信息(包括表面形状)是至关重要的。在这方面,我们提出了一种新的理论和计算算法,旨在定义一组由空间中不均匀分布的点组成的分子表面,以便通过最小化总点数来最大化分子整体形状的信息量。我们通过最近基于二维 Zernike 多项式形式主义的方法来描述分子表面部分的局部形状特性,来测试我们的程序识别热点的能力。这项工作的结果表明,与使用表面上所有点进行描述的完整表面相比,该算法能够使用较少的点来保留分子表面的关键信息。事实上,该方法与均匀随机采样相比,在采样过程中存储的信息方面有显著的提高。