Previtali Viola, Mihigo Helene B, Amet Rebecca, McElligott Anthony M, Zisterer Daniela M, Rozas Isabel
School of Chemistry, Trinity Biomedical Sciences Institute (TBSI), Trinity College Dublin (TCD), 152-160 Pearse Street, D02R590 Dublin 2, Ireland.
School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute (TBSI), Trinity College Dublin (TCD), 152-160 Pearse Street, D02R590 Dublin 2, Ireland.
Pharmaceuticals (Basel). 2020 Dec 21;13(12):485. doi: 10.3390/ph13120485.
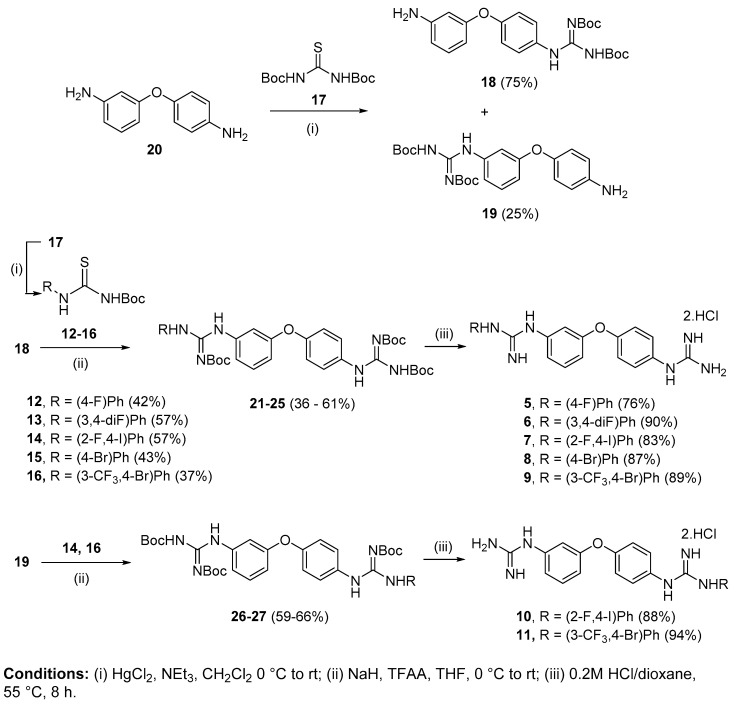
We previously identified a guanidinium-based lead compound that inhibited BRAF through a hypothetic type-III allosteric mechanism. Considering the pharmacophore identified in this lead compound (i.e., "lipophilic group", "di-substituted guanidine", "phenylguanidine polar end"), several modifications were investigated to improve its cytotoxicity in different cancer cell lines. Thus, several were explored, the was replaced by a secondary amine and the phenyl ring in the was substituted by a pyridine. In a structure-based design approach, four representative derivatives were docked into an in-house model of an active triphosphate-containing BRAF protein, and the interactions established were analysed. Based on these computational studies, a variety of derivatives was synthesized, and their predicted drug-like properties calculated. Next, the effect on cell viability of these compounds was assessed in cell line models of promyelocytic leukaemia and breast, cervical and colorectal carcinomas. The potential of a selection of these compounds as apoptotic agents was assessed by screening in the promyelocytic leukaemia cell line HL-60. The toxicity against non-tumorigenic epithelial MCF10A cells was also investigated. These studies allowed for several structure-activity relationships to be derived. Investigations on the mechanism of action of representative compounds suggest a divergent effect on inhibition of the MAPK/ERK signalling pathway.
我们之前鉴定出一种基于胍鎓的先导化合物,它通过一种假设的III型变构机制抑制BRAF。考虑到在这种先导化合物中鉴定出的药效团(即“亲脂性基团”、“二取代胍”、“苯基胍极性端”),我们研究了几种修饰方法以提高其在不同癌细胞系中的细胞毒性。因此,我们探索了几种方法,将胍的[此处原文缺失部分信息]替换为仲胺,并将[此处原文缺失部分信息]中的苯环替换为吡啶。在基于结构的设计方法中,将四种代表性衍生物对接至含活性三磷酸的BRAF蛋白的内部模型中,并分析所建立的相互作用。基于这些计算研究,合成了多种衍生物,并计算了它们预测的类药性质。接下来,在早幼粒细胞白血病以及乳腺癌、宫颈癌和结直肠癌的细胞系模型中评估了这些化合物对细胞活力的影响。通过在早幼粒细胞白血病细胞系HL-60中进行筛选,评估了所选这些化合物作为凋亡剂的潜力。还研究了它们对非致瘤性上皮MCF10A细胞的毒性。这些研究得出了几种构效关系。对代表性化合物作用机制的研究表明,它们对MAPK/ERK信号通路抑制的影响存在差异。