Department of Biology, University of Texas at Arlington, Arlington, Texas, United States of America.
Department of Biochemistry & Molecular Genetics, University of Colorado School of Medicine, Aurora, Colorado, United States of America.
PLoS Negl Trop Dis. 2021 Jan 6;15(1):e0009020. doi: 10.1371/journal.pntd.0009020. eCollection 2021 Jan.
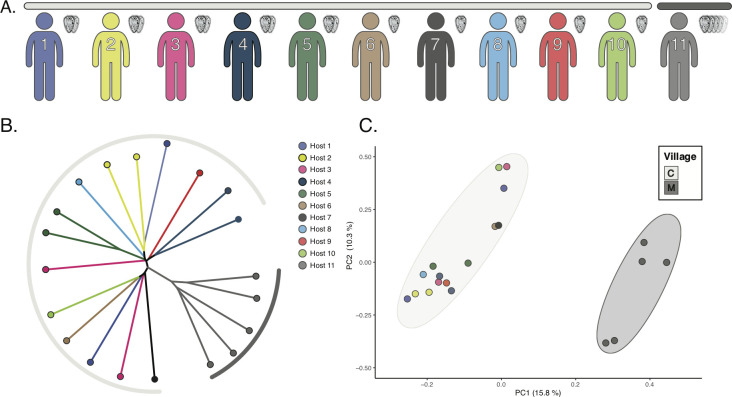
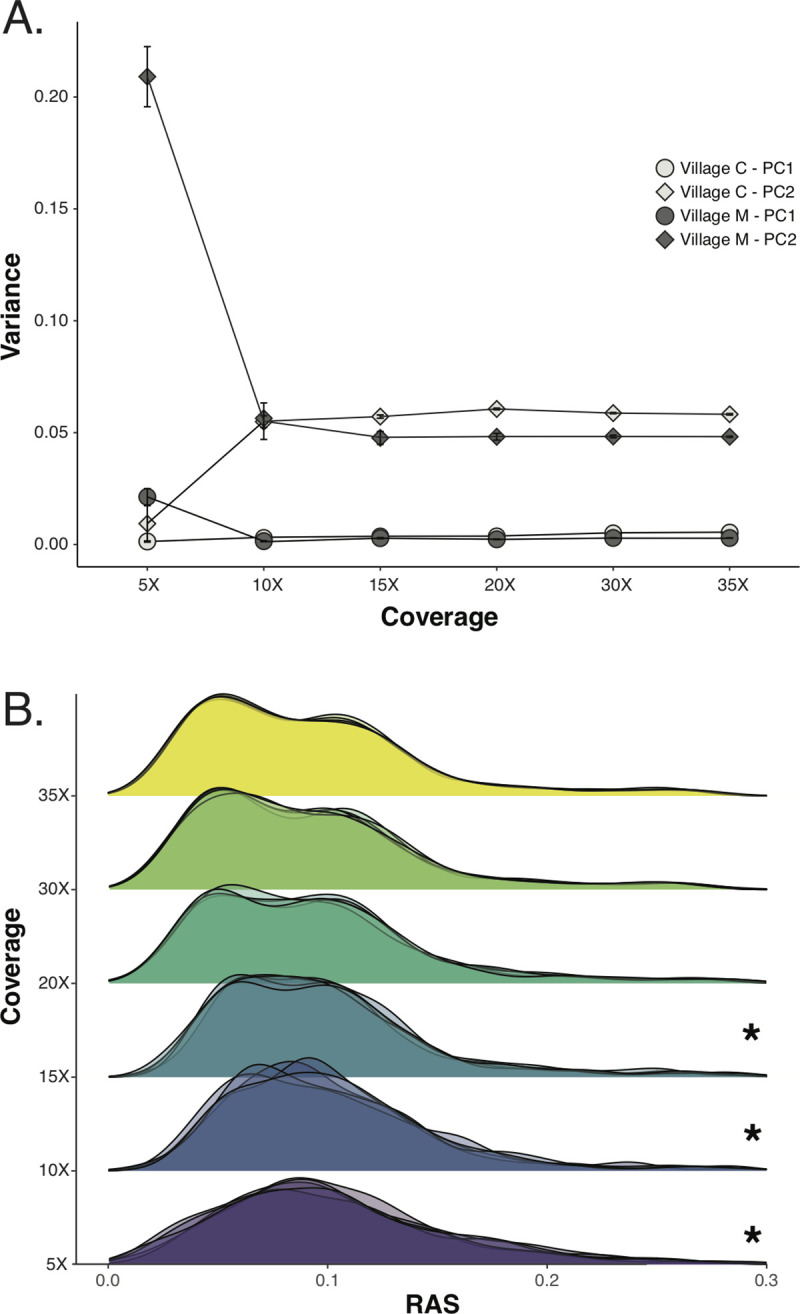
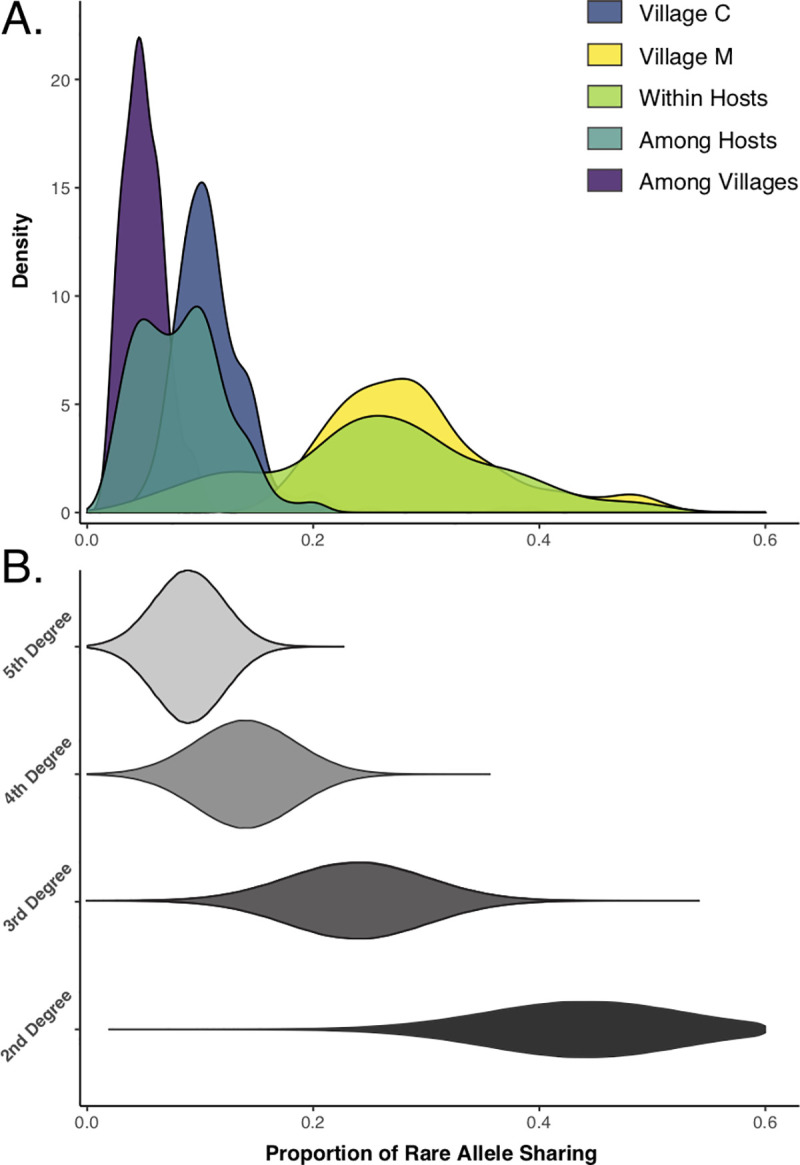
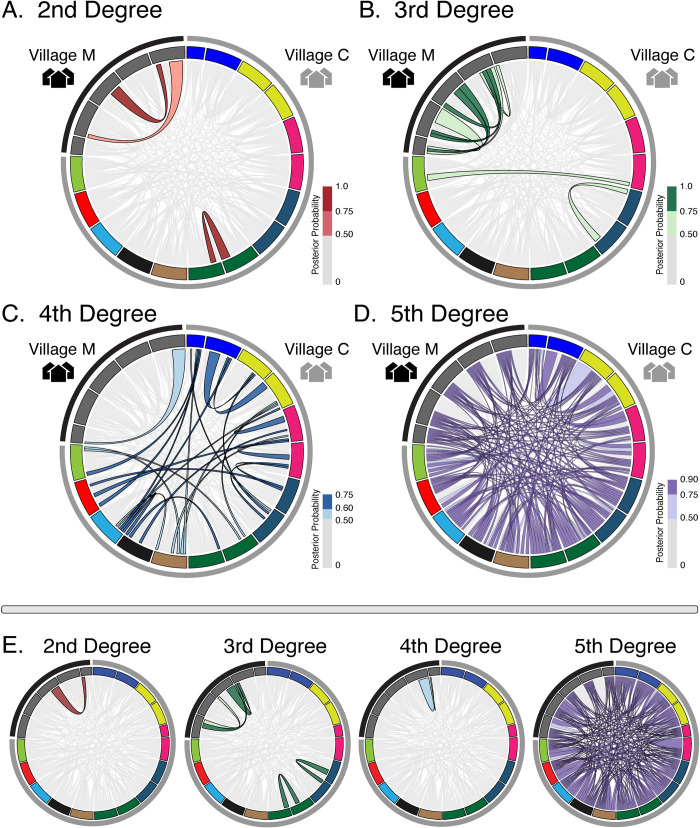
Genomic approaches hold great promise for resolving unanswered questions about transmission patterns and responses to control efforts for schistosomiasis and other neglected tropical diseases. However, the cost of generating genomic data and the challenges associated with obtaining sufficient DNA from individual schistosome larvae (miracidia) from mammalian hosts have limited the application of genomic data for studying schistosomes and other complex macroparasites. Here, we demonstrate the feasibility of utilizing whole genome amplification and sequencing (WGS) to analyze individual archival miracidia. As an example, we sequenced whole genomes of 22 miracidia from 11 human hosts representing two villages in rural Sichuan, China, and used these data to evaluate patterns of relatedness and genetic diversity. We also down-sampled our dataset to test how lower coverage sequencing could increase the cost effectiveness of WGS while maintaining power to accurately infer relatedness. Collectively, our results illustrate that population-level WGS datasets are attainable for individual miracidia and represent a powerful tool for ultimately providing insight into overall genetic diversity, parasite relatedness, and transmission patterns for better design and evaluation of disease control efforts.
基因组学方法为解决血吸虫病和其他被忽视的热带病的传播模式和对控制措施的反应方面的未解决问题提供了巨大的希望。然而,生成基因组数据的成本以及从哺乳动物宿主中获得足够的单个血吸虫幼虫(尾蚴)DNA 的相关挑战限制了基因组数据在研究血吸虫和其他复杂的大型寄生虫中的应用。在这里,我们证明了利用全基因组扩增和测序(WGS)来分析单个存档尾蚴的可行性。作为一个例子,我们对来自中国四川省两个农村村庄的 11 个人类宿主的 22 个尾蚴进行了全基因组测序,并使用这些数据评估了亲缘关系和遗传多样性的模式。我们还对我们的数据集进行了下采样,以测试在保持准确推断亲缘关系的能力的同时,更低的测序覆盖率如何提高 WGS 的成本效益。总的来说,我们的结果表明,个体尾蚴的群体水平 WGS 数据集是可以实现的,这是一种强大的工具,最终可以提供对整体遗传多样性、寄生虫亲缘关系和传播模式的深入了解,从而更好地设计和评估疾病控制措施。