Saka Ryuta, Yamamoto Dan, Kuroda Seika, Ibuka Souji, Kodama Tasuku, Hasegawa Toshimichi
Department of Pediatric Surgery, National Hospital Organization Fukuyama Medical Center, 4-14-17 Okinogamicho, Fukuyama, Hiroshima, 720-8520, Japan.
Department of Obstetrics and Gynecology, National Hospital Organization Fukuyama Medical Center, 4-14-17 Okinogamicho, Fukuyama, Hiroshima, 720-8520, Japan.
Surg Case Rep. 2021 Jan 6;7(1):9. doi: 10.1186/s40792-020-01096-1.
Congenital pyloric atresia (CPA) is a rare gastrointestinal anomaly frequently associated with epidermolysis bullosa (EB). Although the complications of familial isolated CPA are minor, delays in diagnosis can increase the chances of morbidity.
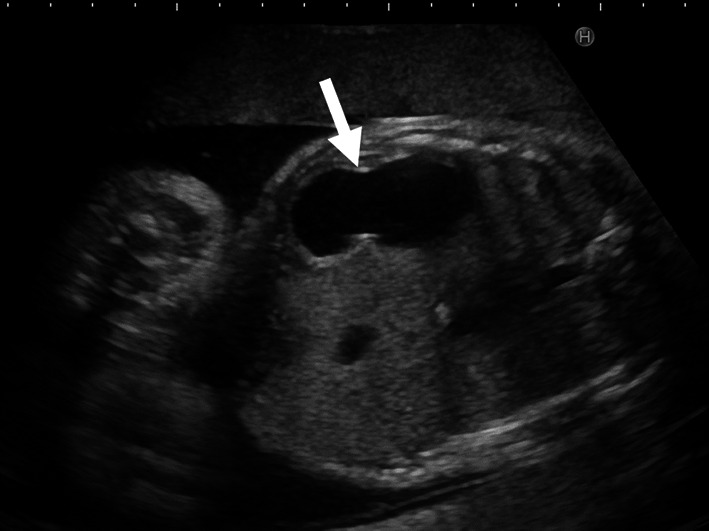
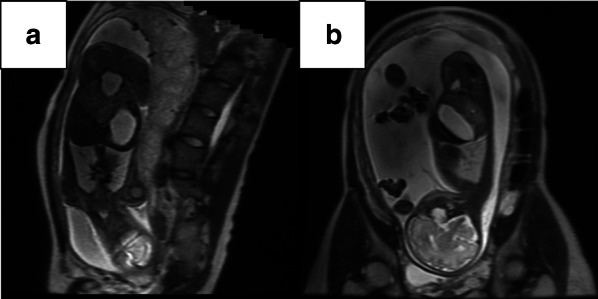
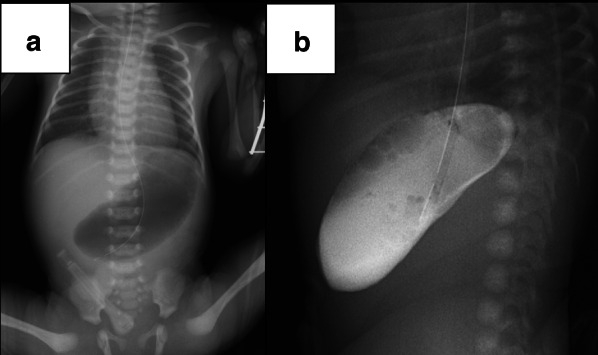
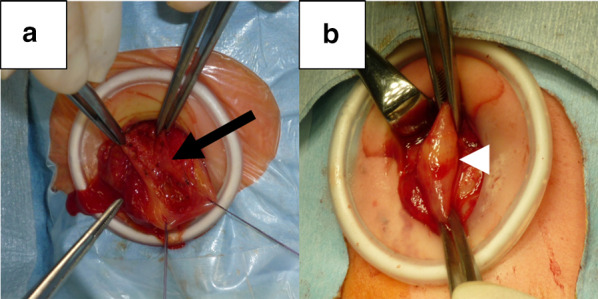
Three female infants born to a Japanese mother presented with CPA at birth. There was no consanguinity between the parents, and the spacing between pregnancies was 2 years in each case. All 3 siblings had a prenatal diagnosis of CPA owing to polyhydramnios and a dilated stomach, without dilatation of the rest of the gastrointestinal tract. All patients underwent reconstructive surgeries for establishing bowel continuity (Case 1, pyloromyotomy; Case 2, gastroduodenostomy in a diamond fashion; and Case 3, gastroduodenostomy in a side-to-side fashion) soon after birth. Their postoperative courses were uneventful, and they grew up healthily, without any complications.
Fetal ultrasonography is useful for diagnosing CPA prenatally. Successful prenatal diagnosis can lead to timely intervention after birth.
先天性幽门闭锁(CPA)是一种罕见的胃肠道畸形,常与大疱性表皮松解症(EB)相关。虽然家族性孤立性CPA的并发症较轻,但诊断延迟会增加发病几率。
一名日本母亲所生的三名女婴出生时即患有CPA。父母无血缘关系,每次怀孕间隔均为2年。由于羊水过多和胃扩张,所有3名兄弟姐妹均在产前被诊断为CPA,胃肠道其他部位未扩张。所有患者出生后不久均接受了重建手术以建立肠道连续性(病例1,幽门肌切开术;病例2,菱形胃十二指肠吻合术;病例3,侧侧胃十二指肠吻合术)。她们术后恢复顺利,健康成长,无任何并发症。
胎儿超声检查有助于产前诊断CPA。成功的产前诊断可导致出生后及时干预。