Kanuri Babunageswararao, Fong Vincent, Ponny Sithara Raju, Tallman Keri A, Rao Sriganesh Ramachandra, Porter Ned, Fliesler Steven J, Patel Shailendra B
Division of Endocrinology, Diabetes and Metabolism, University of Cincinnati, Cincinnati, OH, USA.
Division of Human Genetics, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA.
J Lipid Res. 2021;62:100002. doi: 10.1194/jlr.RA120001101. Epub 2020 Nov 22.
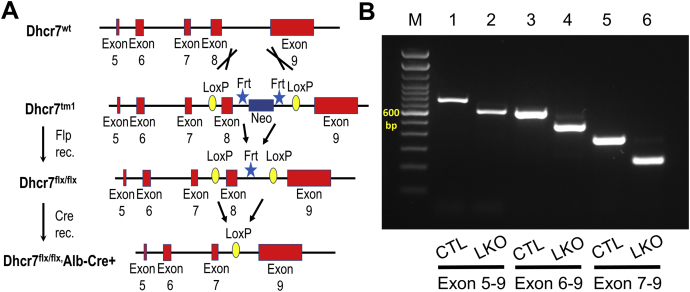
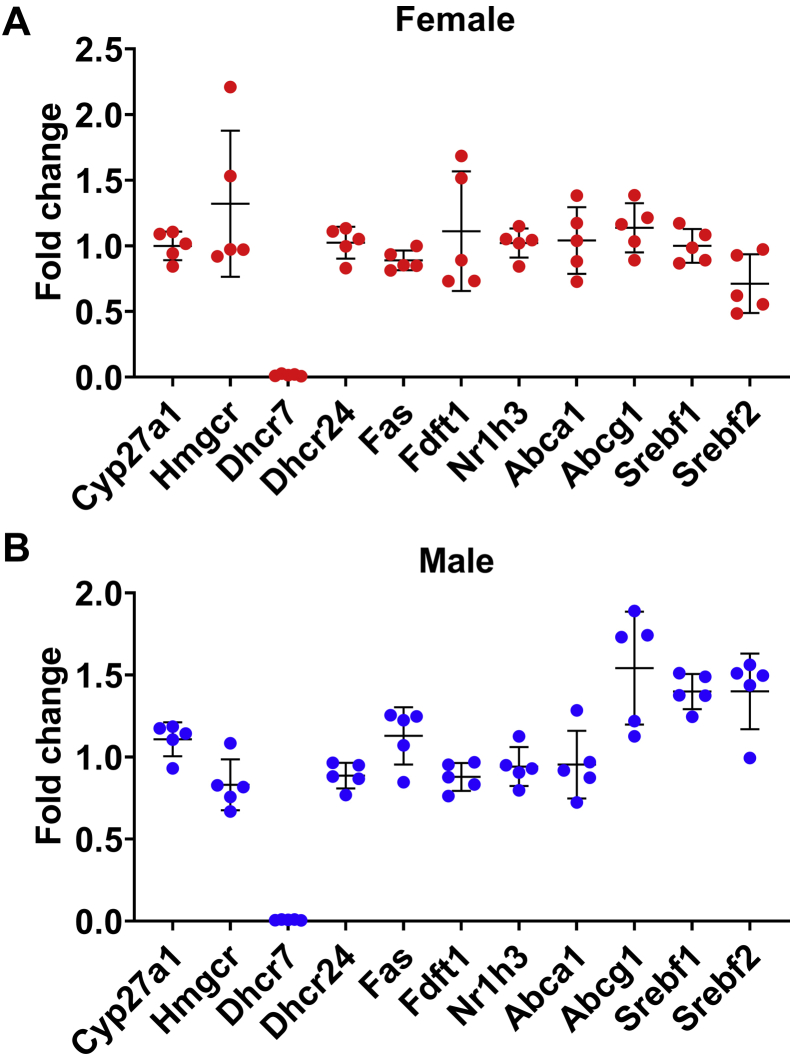
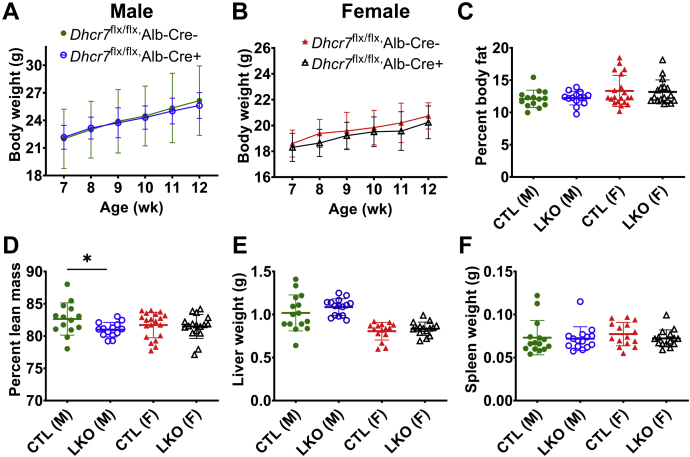
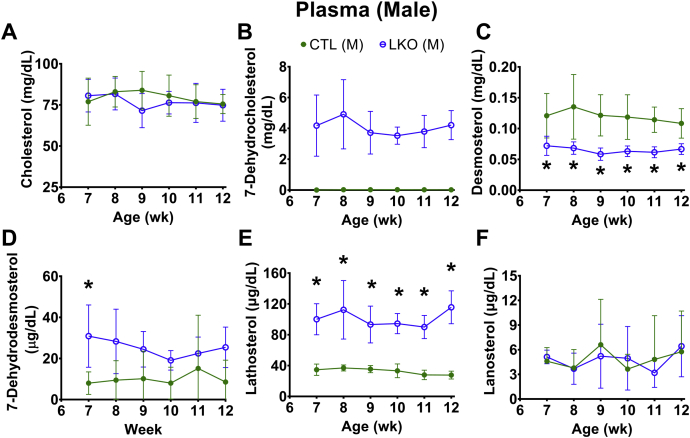
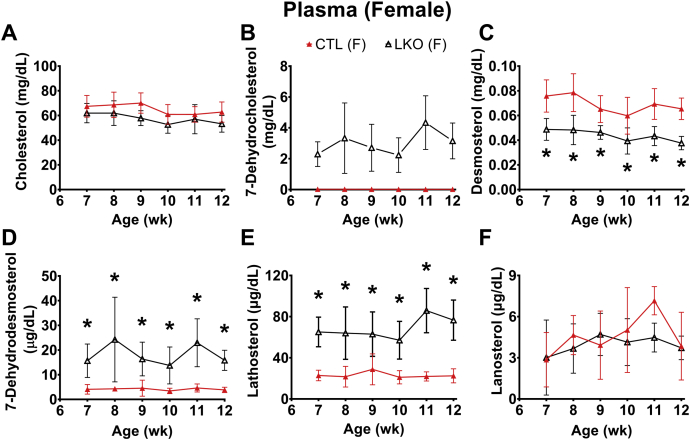
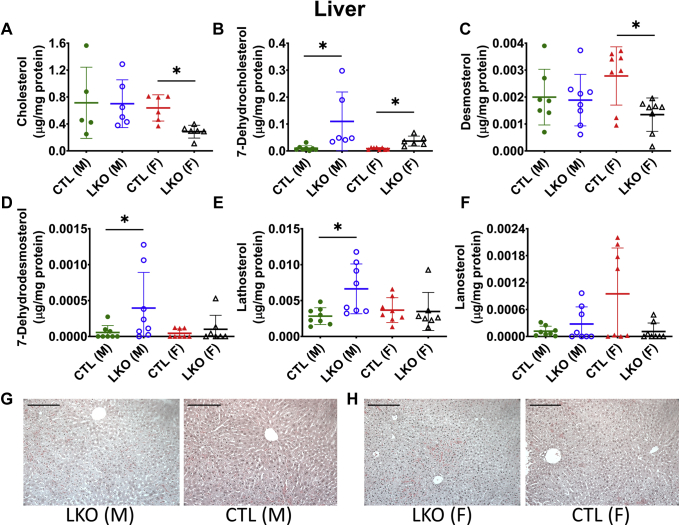
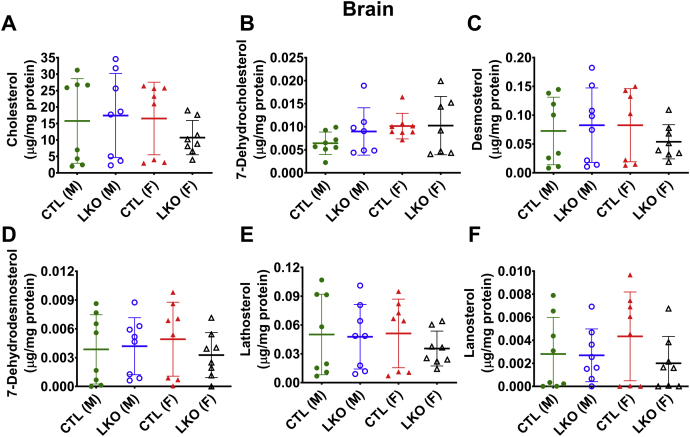
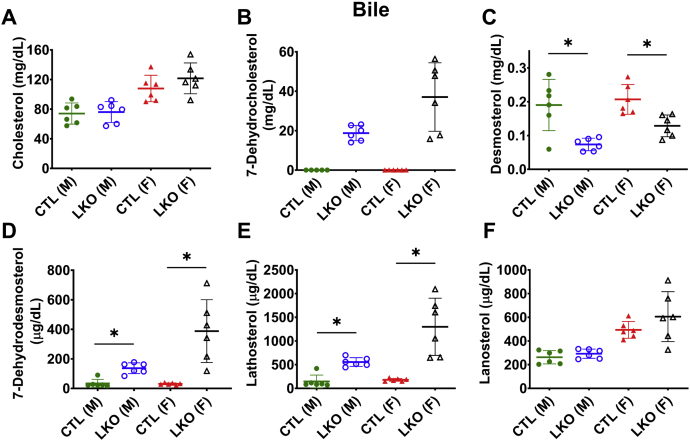
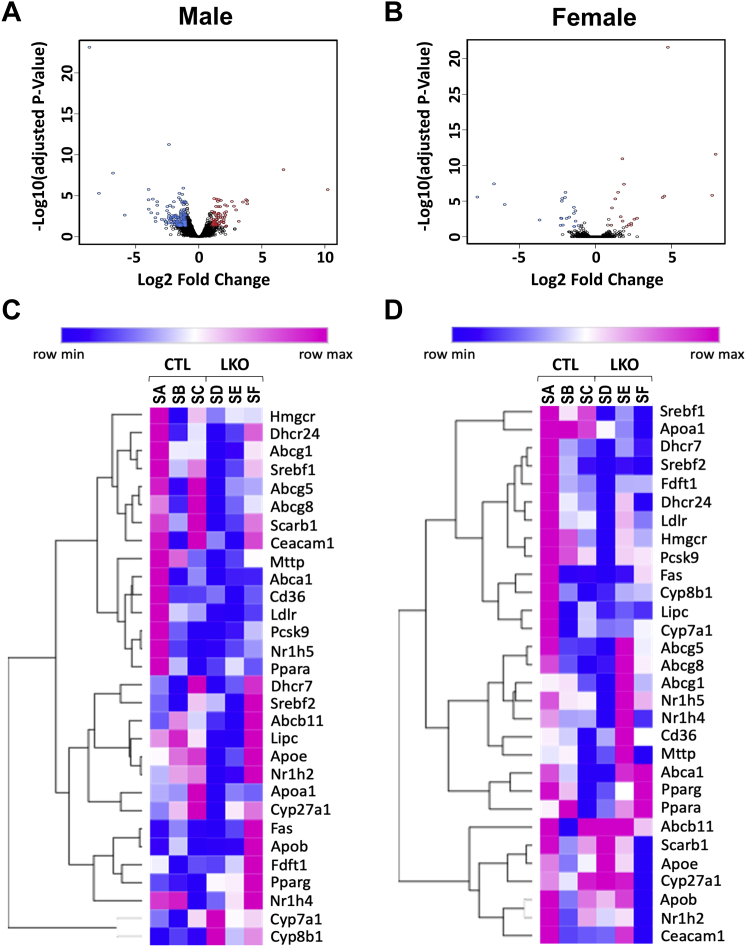
Smith-Lemli-Opitz Syndrome (SLOS) is a developmental disorder (OMIM #270400) caused by autosomal recessive mutations in the Dhcr7 gene, which encodes the enzyme 3β-hydroxysterol-Δ7 reductase. SLOS patients present clinically with dysmorphology and neurological, behavioral, and cognitive defects, with characteristically elevated levels of 7-dehydrocholesterol (7-DHC) in all bodily tissues and fluids. Previous mouse models of SLOS have been hampered by postnatal lethality when Dhcr7 is knocked out globally, while a hypomorphic mouse model showed improvement in the biochemical phenotype with aging and did not manifest most other characteristic features of SLOS. We report the generation of a conditional knockout of Dhcr7 (Dhcr7), validated by generating a mouse with a liver-specific deletion (Dhcr7). Phenotypic characterization of liver-specific knockout mice revealed no significant changes in viability, fertility, growth curves, liver architecture, hepatic triglyceride secretion, or parameters of systemic glucose homeostasis. Furthermore, qPCR and RNA-Seq analyses of livers revealed no perturbations in pathways responsible for cholesterol synthesis, either in male or in female Dhcr7 mice, suggesting that hepatic disruption of postsqualene cholesterol synthesis leads to minimal impact on sterol metabolism in the liver. This validated conditional Dhcr7 knockout model may now allow us to systematically explore the pathophysiology of SLOS, by allowing for temporal, cell and tissue-specific loss of DHCR7.
史密斯-勒米-奥皮茨综合征(SLOS)是一种发育障碍(OMIM #270400),由Dhcr7基因的常染色体隐性突变引起,该基因编码3β-羟基甾醇-Δ7还原酶。SLOS患者临床上表现为形态异常以及神经、行为和认知缺陷,其所有身体组织和体液中的7-脱氢胆固醇(7-DHC)水平均有特征性升高。先前的SLOS小鼠模型因全局敲除Dhcr7后出现出生后致死性而受到阻碍,而一个低表达小鼠模型随着年龄增长其生化表型有所改善,且未表现出SLOS的大多数其他特征。我们报告了Dhcr7条件性敲除小鼠(Dhcr7)的产生,并通过构建肝脏特异性缺失的小鼠(Dhcr7)进行了验证。肝脏特异性敲除小鼠的表型特征显示,其活力、生育能力、生长曲线、肝脏结构、肝脏甘油三酯分泌或全身葡萄糖稳态参数均无显著变化。此外,对肝脏进行的qPCR和RNA测序分析表明,无论是雄性还是雌性Dhcr7小鼠,负责胆固醇合成的途径均未受到干扰,这表明角鲨烯后胆固醇合成的肝脏破坏对肝脏中的甾醇代谢影响极小。这种经过验证的Dhcr7条件性敲除模型现在可能使我们能够通过允许DHCR7在时间、细胞和组织特异性方面的缺失,系统地探索SLOS的病理生理学。