Glavatskikh Marta, Leguy Jules, Hunault Gilles, Cauchy Thomas, Da Mota Benoit
LERIA, University of Angers, 2 Bd Lavoisier, 49045, Angers, France.
Laboratoire MOLTECH-Anjou, UMR CNRS 6200, SFR MATRIX, UNIV Angers, 2 Bd Lavoisier, 49045, Angers, France.
J Cheminform. 2019 Nov 12;11(1):69. doi: 10.1186/s13321-019-0391-2.
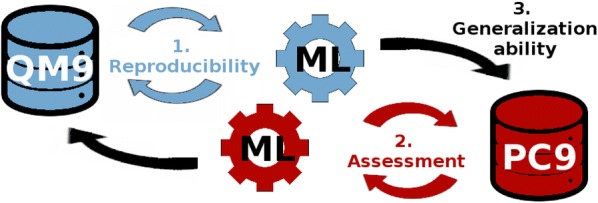
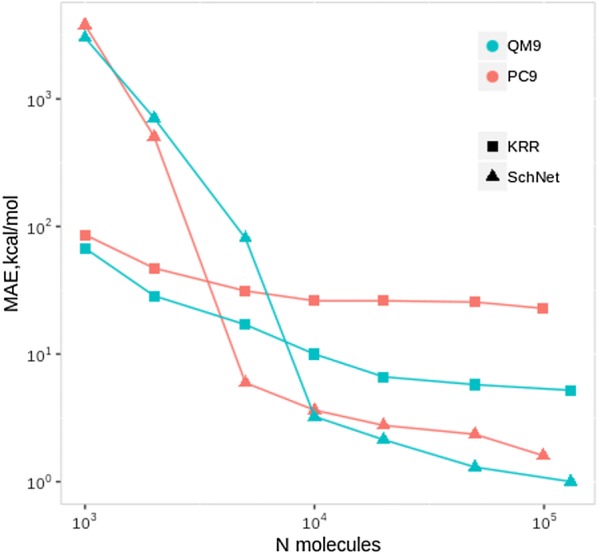
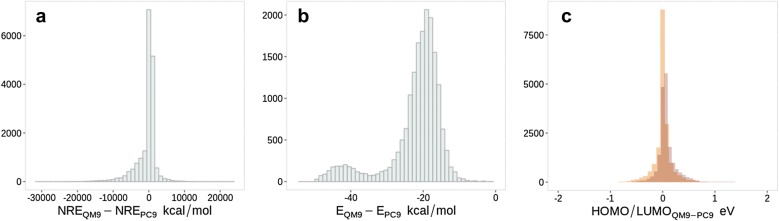
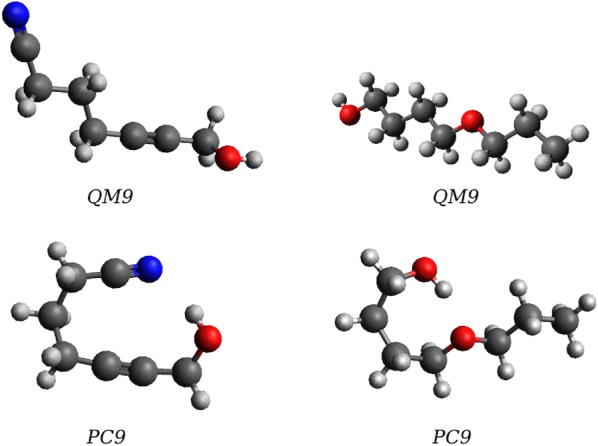
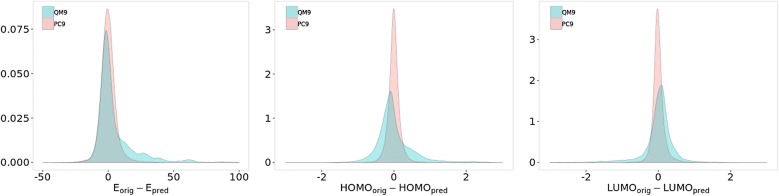
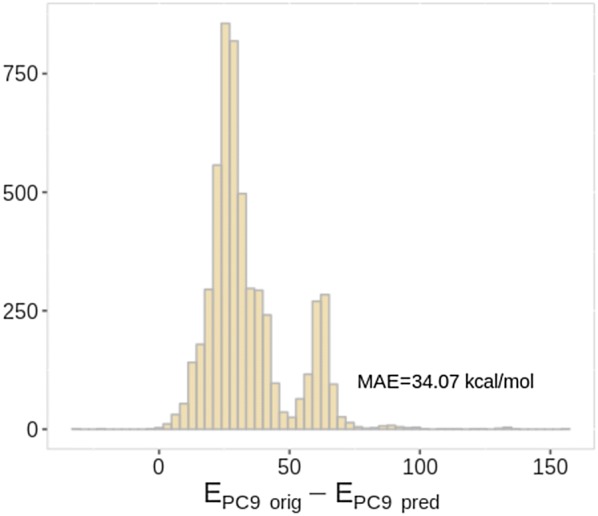
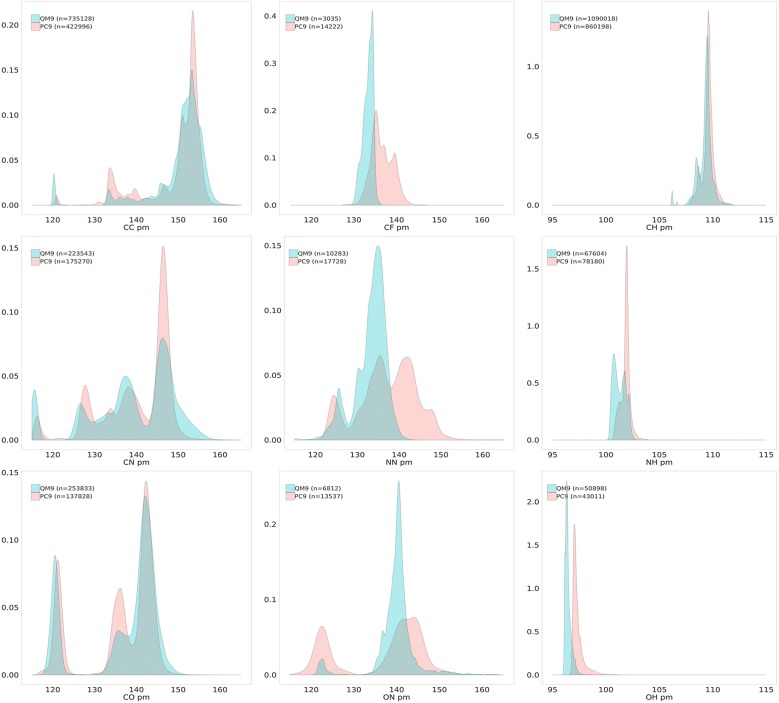
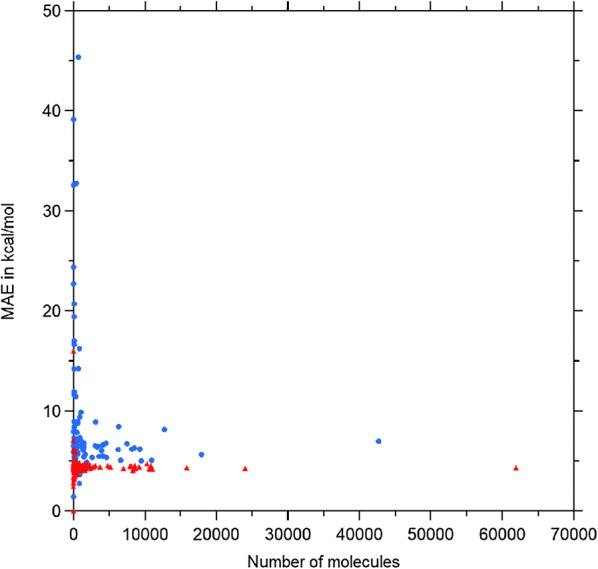
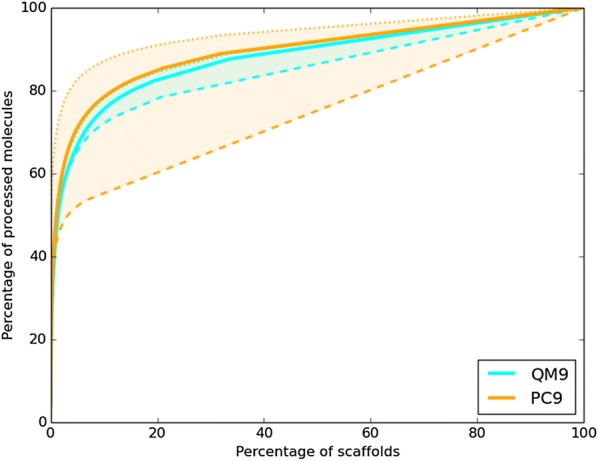
The QM9 dataset has become the golden standard for Machine Learning (ML) predictions of various chemical properties. QM9 is based on the GDB, which is a combinatorial exploration of the chemical space. ML molecular predictions have been recently published with an accuracy on par with Density Functional Theory calculations. Such ML models need to be tested and generalized on real data. PC9, a new QM9 equivalent dataset (only H, C, N, O and F and up to 9 "heavy" atoms) of the PubChemQC project is presented in this article. A statistical study of bonding distances and chemical functions shows that this new dataset encompasses more chemical diversity. Kernel Ridge Regression, Elastic Net and the Neural Network model provided by SchNet have been used on both datasets. The overall accuracy in energy prediction is higher for the QM9 subset. However, a model trained on PC9 shows a stronger ability to predict energies of the other dataset.
QM9数据集已成为机器学习(ML)预测各种化学性质的黄金标准。QM9基于GDB,而GDB是对化学空间的组合探索。最近发表的ML分子预测结果在准确性上与密度泛函理论计算相当。此类ML模型需要在真实数据上进行测试和泛化。本文介绍了PubChemQC项目的一个新的与QM9等效的数据集PC9(仅包含H、C、N、O和F以及最多9个“重”原子)。对键距和化学官能团的统计研究表明,这个新数据集涵盖了更多的化学多样性。在这两个数据集上都使用了核岭回归、弹性网络和SchNet提供的神经网络模型。QM9子集在能量预测方面的总体准确性更高。然而,在PC9上训练的模型在预测另一个数据集的能量方面表现出更强的能力。