Laboratory of Molecular PharmacokineticsGraduate School of Pharmaceutical SciencesUniversity of TokyoTokyoJapan.
Department of Pediatrics and NeonatologyNagoya City University Graduate School of Medical SciencesNagoyaJapan.
Hepatol Commun. 2020 Sep 26;5(1):52-62. doi: 10.1002/hep4.1605. eCollection 2021 Jan.
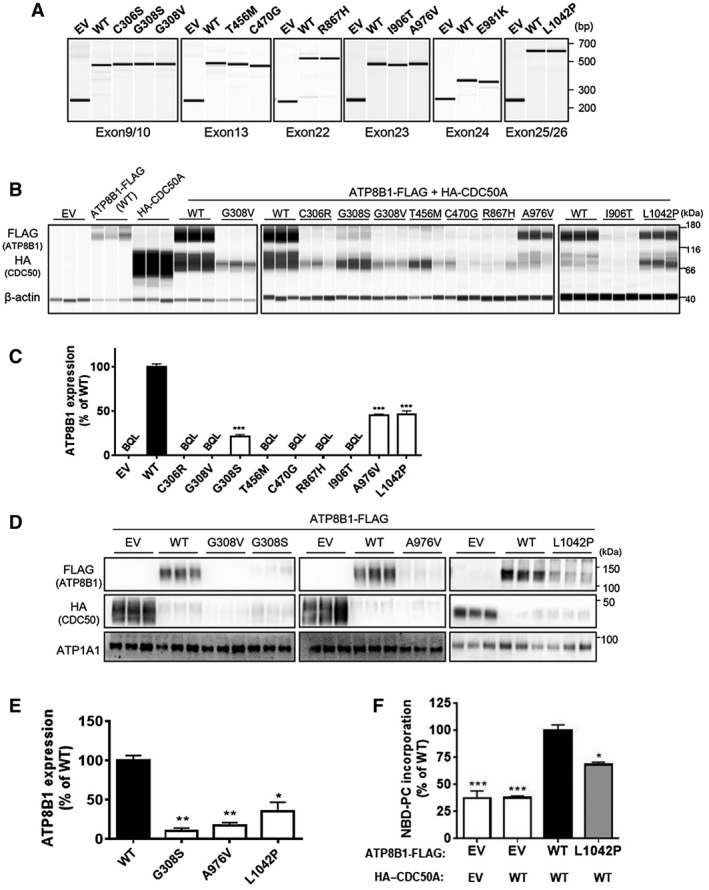
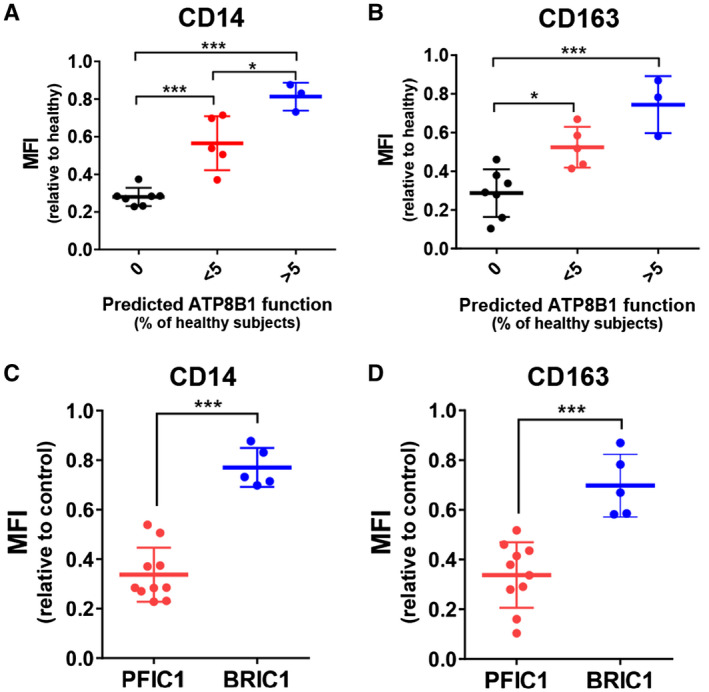
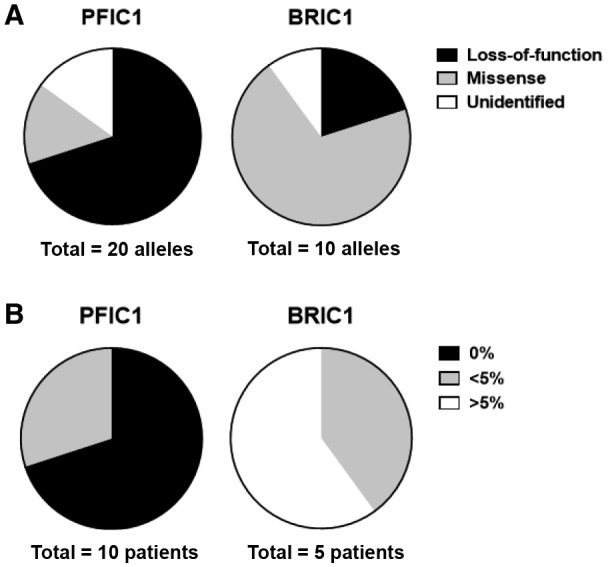
Adenosine triphosphatase phospholipid transporting 8B1 (ATP8B1) deficiency, an ultrarare autosomal recessive liver disease, includes severe and mild clinical forms, referred to as progressive familial intrahepatic cholestasis type 1 (PFIC1) and benign recurrent intrahepatic cholestasis type 1 (BRIC1), respectively. There is currently no practical method for determining PFIC1 or BRIC1 at an early disease course phase. Herein, we assessed the feasibility of developing a diagnostic method for PFIC1 and BRIC1. A nationwide Japanese survey conducted since 2015 identified 25 patients with cholestasis with mutations, 15 of whom agreed to participate in the study. Patients were divided for analysis into PFIC1 (n = 10) or BRIC1 (n = 5) based on their disease course. An mutagenesis assay to evaluate pathogenicity of mutations suggested that residual ATP8B1 function in the patients could be used to identify clinical course. To assess their ATP8B1 function more simply, human peripheral blood monocyte-derived macrophages (HMDMs) were prepared from each patient and elicited into a subset of alternatively activated macrophages (M2c) by interleukin-10 (IL-10). This was based on our previous finding that ATP8B1 contributes to polarization of HMDMs into M2c. Flow cytometric analysis showed that expression of M2c-related surface markers cluster of differentiation (CD)14 and CD163 were 2.3-fold and 2.1-fold lower (95% confidence interval, 2.0-2.5 for CD14 and 1.7-2.4 for CD163), respectively, in patients with IL-10-treated HMDMs from PFIC1 compared with BRIC1. : CD14 and CD163 expression levels in IL-10-treated HMDMs may facilitate diagnosis of PFIC1 or BRIC1 in patients with ATP8B1 deficiency.
三磷酸腺苷酶磷脂转运蛋白 8B1(ATP8B1)缺乏症是一种极罕见的常染色体隐性肝脏疾病,包括严重和轻度的临床表现,分别称为进行性家族性肝内胆汁淤积症 1 型(PFIC1)和良性复发性肝内胆汁淤积症 1 型(BRIC1)。目前尚无在疾病早期阶段确定 PFIC1 或 BRIC1 的实用方法。在此,我们评估了开发 PFIC1 和 BRIC1 诊断方法的可行性。自 2015 年以来,一项全国性的日本调查确定了 25 名患有 突变的胆汁淤积患者,其中 15 名同意参与该研究。根据疾病过程,患者被分为 PFIC1(n=10)或 BRIC1(n=5)进行分析。评估 突变致病性的突变基因检测表明,患者中残留的 ATP8B1 功能可用于确定临床过程。为了更简单地评估他们的 ATP8B1 功能,从每位患者中制备了人外周血单核细胞衍生的巨噬细胞(HMDM),并通过白细胞介素-10(IL-10)将其诱导为一组替代性激活的巨噬细胞(M2c)。这是基于我们之前的发现,即 ATP8B1 有助于 HMDM 向 M2c 极化。流式细胞术分析显示,与 BRIC1 相比,IL-10 处理的来自 PFIC1 的 HMDM 中 M2c 相关表面标志物 CD14 和 CD163 的表达分别低 2.3 倍和 2.1 倍(CD14 的 95%置信区间为 2.0-2.5,CD163 的为 1.7-2.4)。结论:IL-10 处理的 HMDM 中 CD14 和 CD163 的表达水平可能有助于 ATP8B1 缺乏症患者中 PFIC1 或 BRIC1 的诊断。