Bioinformatics and Systems Biology Program, University of California San Diego, La Jolla, CA, USA.
Department of Bioengineering, University of California San Diego, La Jolla, CA, USA.
Nature. 2021 Jan;589(7841):246-250. doi: 10.1038/s41586-020-03078-7. Epub 2021 Jan 13.
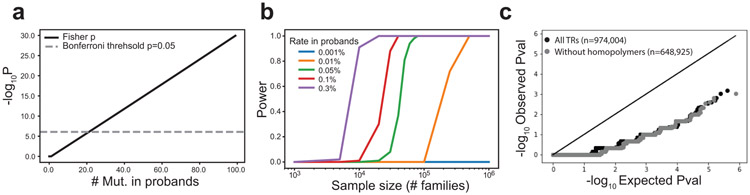
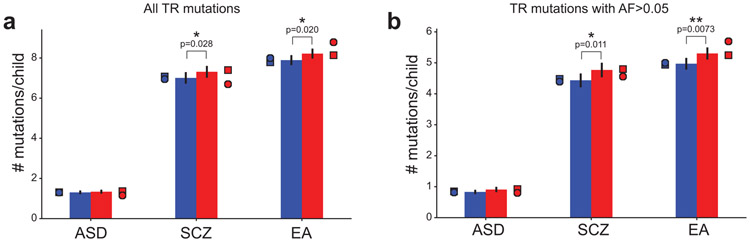
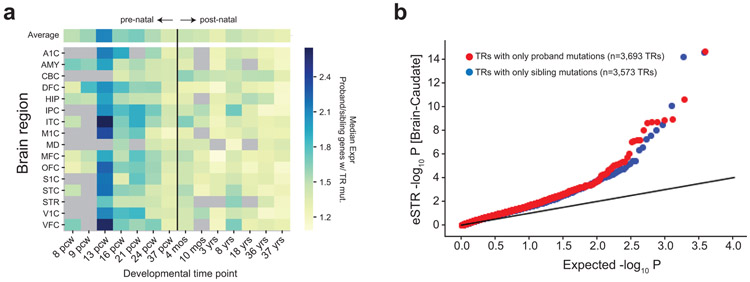
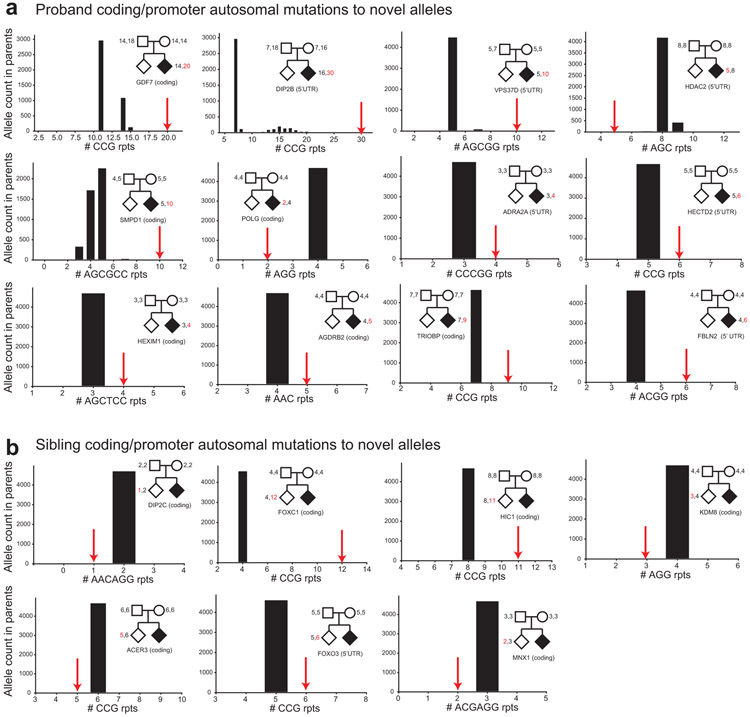
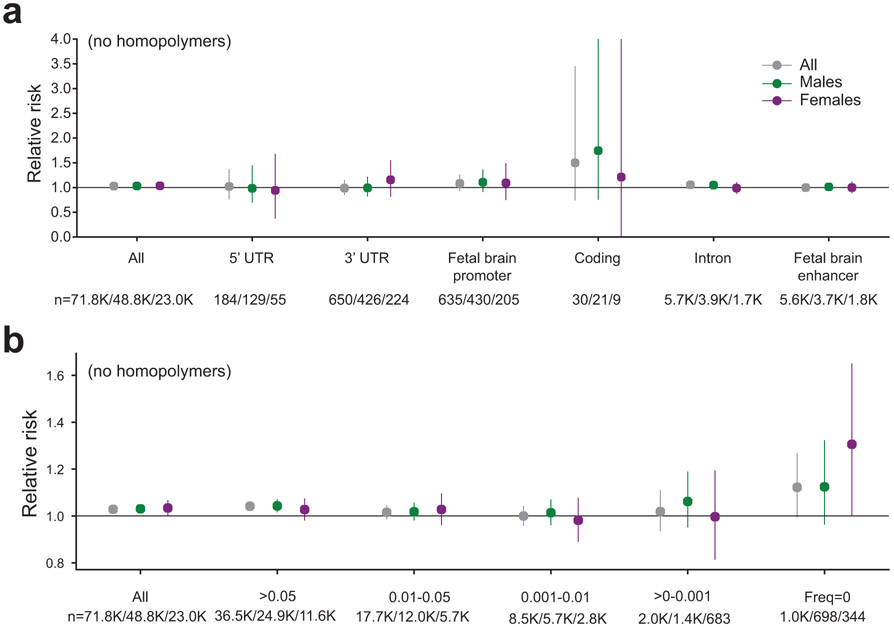
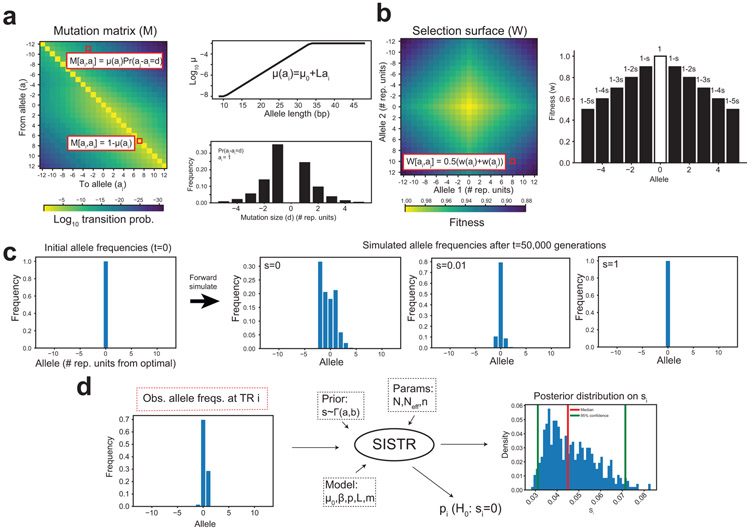
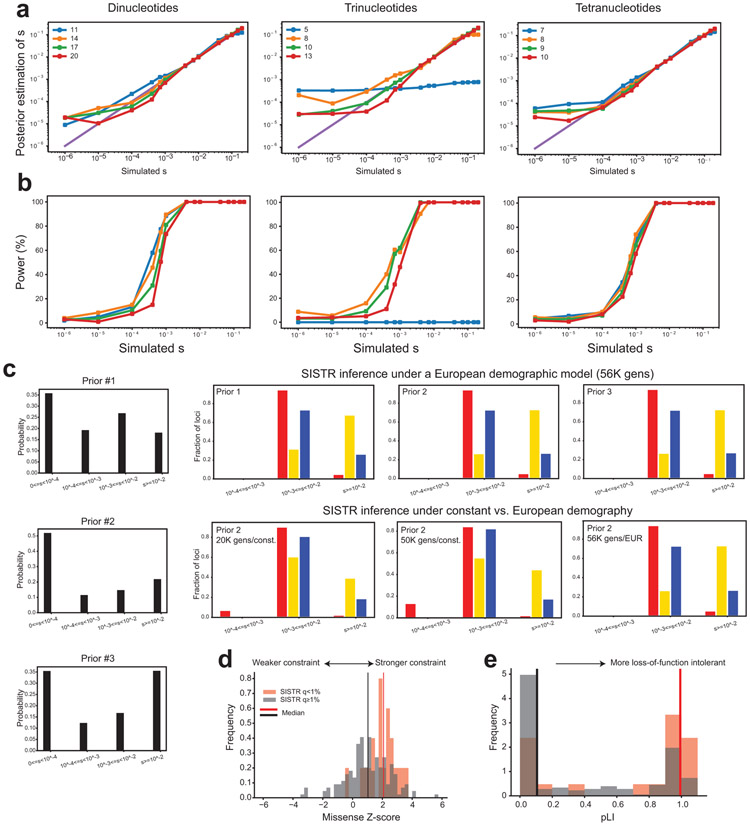
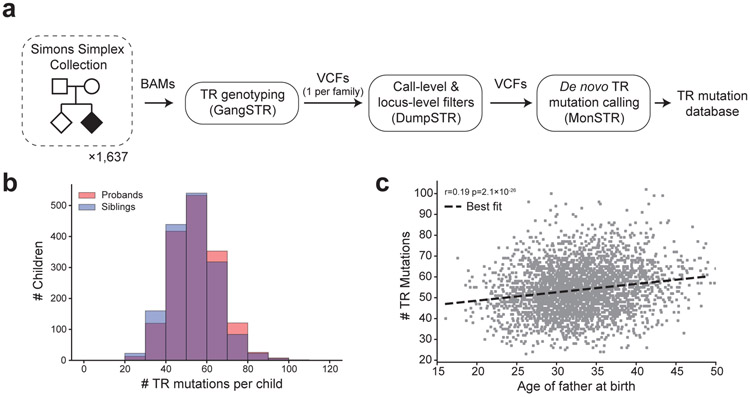
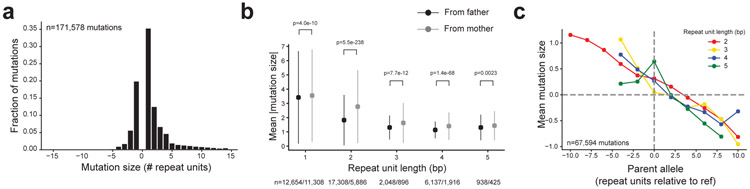
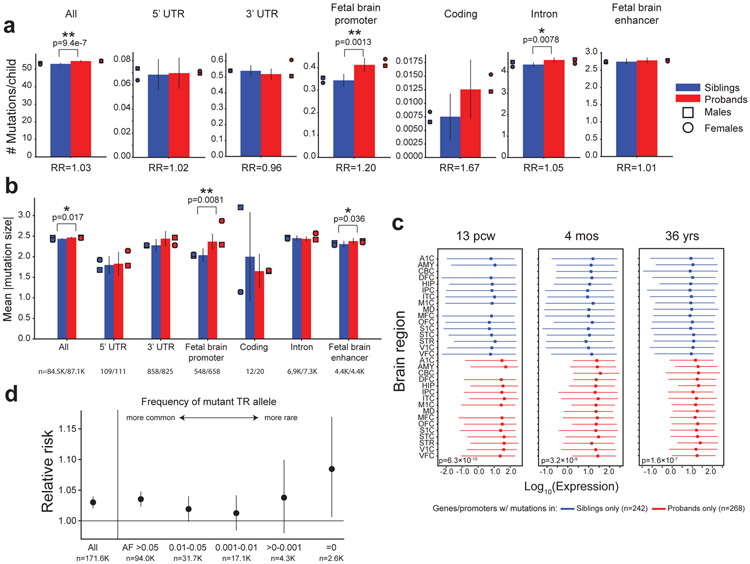
Autism spectrum disorder (ASD) is an early-onset developmental disorder characterized by deficits in communication and social interaction and restrictive or repetitive behaviours. Family studies demonstrate that ASD has a substantial genetic basis with contributions both from inherited and de novo variants. It has been estimated that de novo mutations may contribute to 30% of all simplex cases, in which only a single child is affected per family. Tandem repeats (TRs), defined here as sequences of 1 to 20 base pairs in size repeated consecutively, comprise one of the major sources of de novo mutations in humans. TR expansions are implicated in dozens of neurological and psychiatric disorders. Yet, de novo TR mutations have not been characterized on a genome-wide scale, and their contribution to ASD remains unexplored. Here we develop new bioinformatics methods for identifying and prioritizing de novo TR mutations from sequencing data and perform a genome-wide characterization of de novo TR mutations in ASD-affected probands and unaffected siblings. We infer specific mutation events and their precise changes in repeat number, and primarily focus on more prevalent stepwise copy number changes rather than large expansions. Our results demonstrate a significant genome-wide excess of TR mutations in ASD probands. Mutations in probands tend to be larger, enriched in fetal brain regulatory regions, and are predicted to be more evolutionarily deleterious. Overall, our results highlight the importance of considering repeat variants in future studies of de novo mutations.
自闭症谱系障碍 (ASD) 是一种早期发病的发育障碍,其特征是沟通和社交互动方面的缺陷,以及限制性行为或重复性行为。家族研究表明,ASD 具有很大的遗传基础,既有遗传的也有新生的变异。据估计,新生突变可能导致所有单纯病例的 30%,即每个家庭只有一个孩子受到影响。串联重复 (TR) 在这里被定义为大小为 1 到 20 个碱基对的序列连续重复,是人类新生突变的主要来源之一。TR 扩展与数十种神经和精神疾病有关。然而,尚未在全基因组范围内对新生 TR 突变进行特征描述,其对 ASD 的贡献仍未得到探索。在这里,我们开发了新的生物信息学方法,用于从测序数据中识别和优先考虑新生 TR 突变,并对受影响的 ASD 先证者和未受影响的兄弟姐妹进行全基因组新生 TR 突变特征描述。我们推断特定的突变事件及其重复数的精确变化,主要关注更常见的逐步拷贝数变化,而不是大的扩展。我们的研究结果表明,ASD 先证者中存在显著的全基因组 TR 突变过多。先证者中的突变往往更大,富集在胎儿大脑调控区域,并且预计更具进化上的有害性。总的来说,我们的研究结果强调了在未来的新生突变研究中考虑重复变异的重要性。