Department of Pathology and Laboratory Medicine, University of Wisconsin-Madison, 555 Science Dr, Madison, WI, 53711, USA.
Infection Analytics Program, Kirby Institute for Infection and Immunity, UNSW Sydney, Sydney, NSW, 2052, Australia.
Virol J. 2021 Jan 15;18(1):21. doi: 10.1186/s12985-020-01473-0.
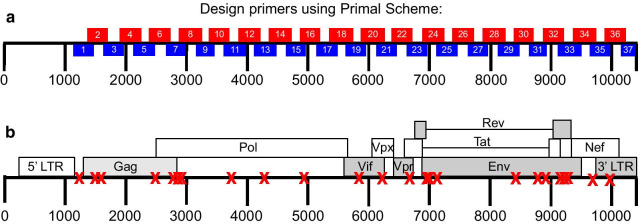
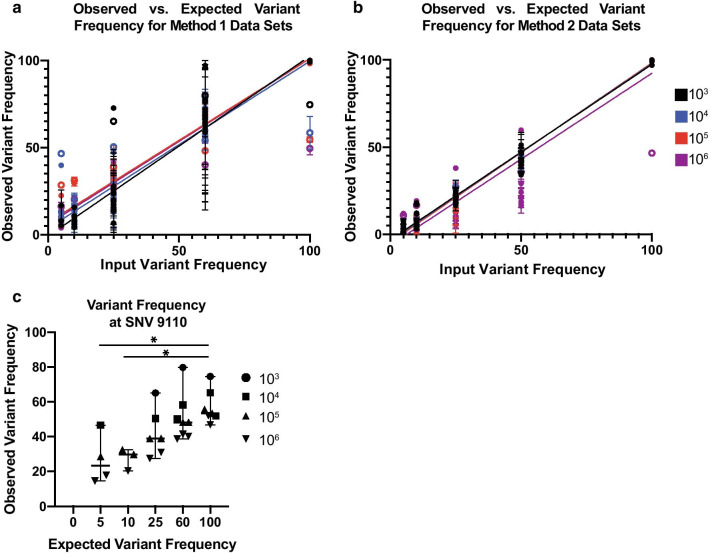
The generation of accurate and reproducible viral sequence data is necessary to understand the diversity present in populations of RNA viruses isolated from clinical samples. While various sequencing methods are available, they often require high quality templates and high viral titer to ensure reliable data.
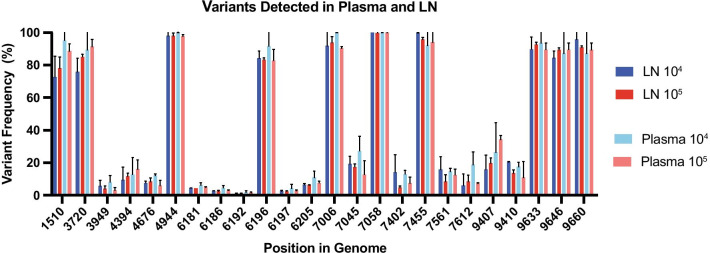
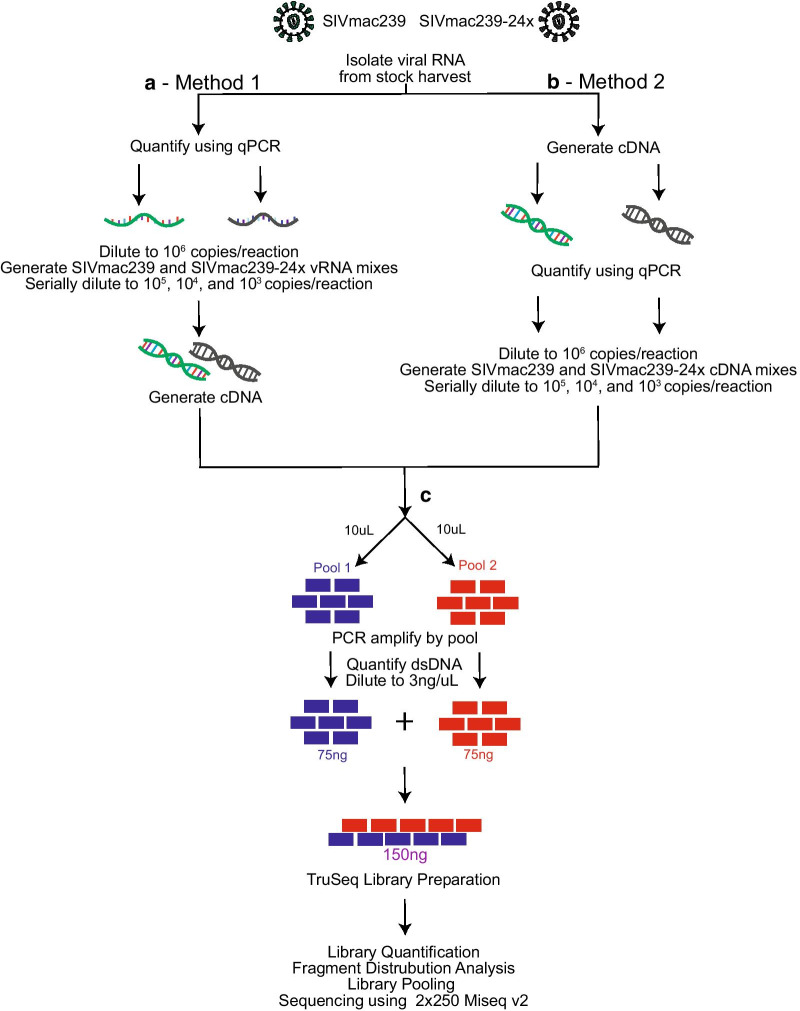
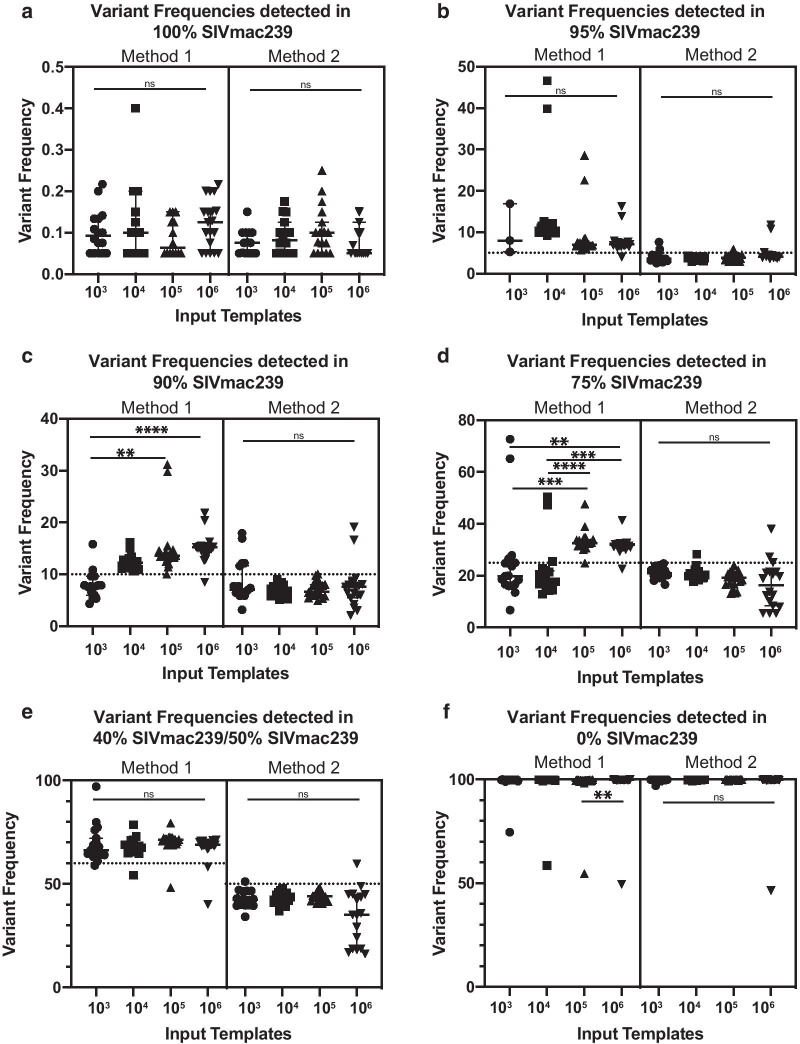
We modified a multiplex PCR and sequencing approach to characterize populations of simian immunodeficiency virus (SIV) isolated from nonhuman primates. We chose this approach with the aim of reducing the number of required input templates while maintaining fidelity and sensitivity. We conducted replicate sequencing experiments using different numbers of quantified viral RNA (vRNA) or viral cDNA as input material. We performed assays with clonal SIVmac239 to detect false positives, and we mixed SIVmac239 and a variant with 24 point mutations (SIVmac239-24X) to measure variant detection sensitivity.
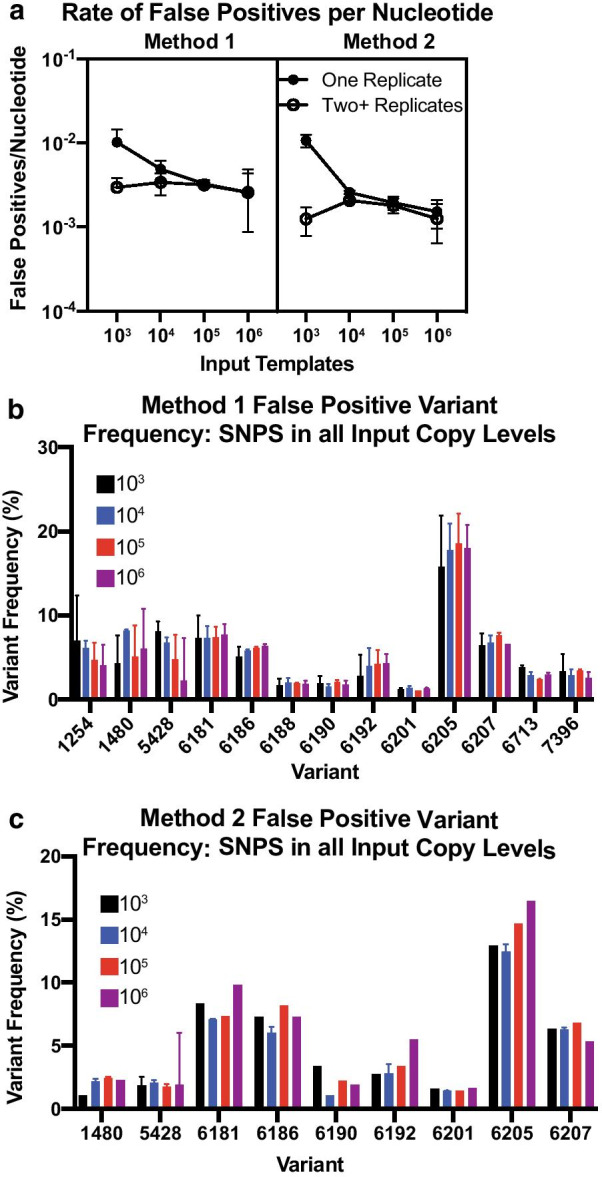
We found that utilizing a starting material of quantified viral cDNA templates had a lower rate of false positives and increased reproducibility when compared to that of quantified vRNA templates. This study identifies the importance of rigorously validating deep sequencing methods and including replicate samples when using a new method to characterize low frequency variants in a population with a small number of templates.
Because the need to generate reproducible and accurate sequencing data from diverse viruses from low titer samples, we modified a multiplex PCR and sequencing approach to characterize SIV from populations from non-human primates. We found that increasing starting template numbers increased the reproducibility and decreased the number of false positives identified, and this was further seen when cDNA was used as a starting material. Ultimately, we highlight the importance of vigorously validating methods to prevent overinterpretation of low frequency variants in a sample.
为了了解从临床样本中分离出的 RNA 病毒群体的多样性,必须生成准确且可重现的病毒序列数据。虽然有多种测序方法,但它们通常需要高质量的模板和高病毒滴度,以确保可靠的数据。
我们修改了多重 PCR 和测序方法,以描述从非人灵长类动物中分离出的猴免疫缺陷病毒(SIV)群体。我们选择这种方法的目的是减少所需输入模板的数量,同时保持保真度和灵敏度。我们使用不同数量的定量病毒 RNA(vRNA)或病毒 cDNA 作为输入材料进行了重复测序实验。我们使用克隆 SIVmac239 进行了检测假阳性的测定,并混合了 SIVmac239 和具有 24 个点突变的变体(SIVmac239-24X),以测量变体检测灵敏度。
我们发现,与定量 vRNA 模板相比,使用定量病毒 cDNA 模板作为起始材料时,假阳性率更低,重现性更高。本研究确定了在使用新方法对模板数量较少的群体中的低频变体进行特征描述时,严格验证深度测序方法并包含重复样本的重要性。
由于需要从低滴度样本中的多种病毒生成可重现且准确的测序数据,我们修改了多重 PCR 和测序方法,以描述来自非人灵长类动物群体的 SIV。我们发现,增加起始模板数量可以提高重现性并减少鉴定出的假阳性数量,当使用 cDNA 作为起始材料时,这种情况更为明显。最终,我们强调了大力验证方法的重要性,以防止对样品中低频变体的过度解释。